
Abstract
Capillaries pervade human physiology. The mean intercapillary distance is only about 100 μm in human tissue, which indicates the extent of nutrient diffusion. In engineered tissue the lack of capillaries, along with the associated perfusion, is problematic because it leads to hypoxic stress and necrosis. However, a capillary is not easy to engineer due to its complex cytoarchitecture. Here, it is shown that it is possible to create in vitro, in about 30 min, a tubular microenvironment with an elastic modulus and porosity consistent with human tissue that functionally mimicks a bona fide capillary using “live cell lithography”(LCL) to control the type and position of cells on a composite hydrogel scaffold. Furthermore, it is established that these constructs support the forces associated with blood flow and produce nutrient gradients similar to those measured in vivo. With LCL, capillaries can be constructed with single cell precision—no other method for tissue engineering offers such precision. Since the time required for assembly scales with the number of cells, this method is likely to be adapted first to create minimal functional units of human tissue that constitute organs, consisting of a heterogeneous population of 100–1000 cells, organized hierarchically to express a predictable function.
Similar content being viewed by others

Introduction
Tissue engineering is a cure. Eventually, tissue may be engineered for organ replacement and regenerative medicine, but in vitro models engineered to accurately emulate human physiology are more likely to have an effect on health sooner, for example, in drug discovery1. In vitro microfluidic organs-on-a-chip offer the promise to recapitulate the minimal functional units (MFU) found in major organs, such as the acinus (liver), nephron (kidney) or alveoli (lung), but so far have failed to reproduce the micro-scale hierarchical organization, spatial heterogeneity and cell-matrix interactions found in organoids or tissue sections2. Responding to these challenges, a number of ways have been developed to co-culture two or more cell types and control the cell positions3,4,5, which include rapid casting5 and bioprinting6,7 and each has advantages and limitations. The prior work has revealed several technical obstacles that still have to be overcome to create functional engineered tissue;1,8 among them are 1. engineered tissue lacks a hierarchical organization required to deliver specific biological functions; 2. the mechanics of the engineered tissue are weak compared to in vivo tissue; 3. a bona fide extracellular matrix (ECM) is cell-specific9, but the scaffolds used to engineer tissue are not; and 4. engineered tissue lacks the microcirculation and the complex vasculature required for perfusion to avoid necrosis—the volume of engineered tissue cannot exceed the diffusion limit10.
“Live cell lithography” (LCL) may possibly resolve these issues. LCL uses multiple optical tweezers to precisely position cells in three-dimensions (3D) in a photo-polymerized hydrogel scaffold—creating “living voxels” with exposed cell surfaces11,12. Furthermore, by stitching the living voxels together, a cytoarchitecture of any size, shape and constituency can be formed and embedded in a microfluidic device for testing. In these constructs, the hydrogel scaffold performs a function similar to the natural ECM, but it is synthetic, sterile and can be hybridized. Importantly, with LCL, it is possible to tailor the microenvironment of each cell-type by photo-polymerizing a specific pre-polymer in each voxel to form cell-specific scaffolds.
As a demonstration, by controlling the type and position of cells on a composite, cell-specific hydrogel scaffold, LCL was used to create perfusable synthetic capillaries in about 30 min. Interestingly, the porosity and stiffness of the cell-specific scaffolds were chosen to test the efficacy of a model for the mechanics that views a capillary as a tunnel through a gel13,14,15. By flowing human blood and oxygenated fluid through it, it was established that these constructs support the forces associated with blood flow and nutrient gradients similar to those measured in vivo in a bona fide human capillary, validating this model for the mechanics.
This is an important test of the prospects for using LCL as a method for tissue engineering. The MFUs in major organs all incorporate capillaries and so this aspect of the cytoarchitecture must be essential to the physiology and represents a first step in the creation of an MFU. However, the complex architecture makes a capillary difficult to engineer repeatably and precisely. Advanced microscopy has revealed its cytoarchitecture16. The lumen of a capillary measures only about 3–30 μm in diameter—so small that erythrocytes (red blood cells) pass through in single file—and it is enclosed by a monolayer of endothelial cells (ECs) that rests on a basement membrane (BM) invested abluminally with pericytes. What is new here is the precision of the cell placement, the control of the cytoarchitecture and the cell-specific scaffolds, delivered by LCL, which may all be instrumental in the development of MFUs. That is not to say that tubular structures resembling capillaries have not been formed in vitro. For example, in tube formation assays, which are widely used as in vitro models for angiogenesis, primary or immortalized ECs, mixed with conditioned media and plated on basement membrane matrix, self-assemble into tubes in about 3–12 hr17, but there is little prospect for creating an MFU this way. On the other hand, LCL is appealing for engineering MFUs because it offers precise control of the cytoarchitecture. As a final illustration of how this precision could be exercised and harnessed to study biology or pathology, cancer cells were repeatedly trapped in synthetic capillaries. This naive model is used to illustrate the advantages and shortcomings of the LCL-approach to tissue engineering.
Results and Discussion
Cell Viability
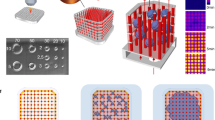
The use of optical tweezers for manipulating living cells offers precise control over the size, shape and composition of the capillary cytoarchitecture (Fig. 1a). However, the concomitant photodamage could compromise cell viability. To avoid absorption, light in the near-infrared (IR) band was employed for tweezing. To assess the effect of the IR radiation wavelength, power and duration of exposure on cell viability, the three cell types used to create a capillary: human microvascular endothelial cells (hMVECs); human pericytes from the placenta (hPPs); and human neonatal fibroblasts (hNFs) were exposed to a time-shared 3×3 array of (nine) optical traps. In pursuit of an optimal wavelength and power, the traps were formed at wavelengths λ = 800, 830, 850 and 870 nm with time-averaged optical power ranging from 0 to 200 mW using a dwell time of 20 μsec over each of nine traps with the duration of exposure to the beam limited to <30 sec. Subsequently, the cells were partially encapsulated in 5.0 kDa PEGDA/PEG-RGDS hydrogel along with dark controls. To promote viability, fresh media flowed through the arrays continuously at a rate of 0.05 μl/min.
The viability scores from this assay remained consistently high (~90%) in the band 830–850 nm for all the cells as long as the time-averaged power ≤ 100 mW; no significant differences were observed between the dark controls (≥ 90% viable) and the number of viable cells after exposure to the trap beam (Fig. 1b). (Likewise MDA-MB-231 breast cancer cells were tested under similar conditions, which has already been reported elsewhere18). The viability was especially sensitive to handling, however (see Methods). Thus, with proper handling, cells trapped at λ = 830 nm for <30 sec in a time-shared trap with a total time-averaged power ≤100 mW remained viable.
Hydrogel Nanomechanics and Composite Scaffolds
Operationally, multiple laminar fluid flows in a four-port microfluidic device were used to convey cell suspensions to a cross-connecting channel where multiple optical tweezers (Fig. 1a, red optical path) were used to pluck them from the flow and assemble them on a hydrogel scaffold. The hydrogel scaffold that partially encapsulated each cell was formed by photopolymerizing a pre-polymer mixture in the cell suspension. Depending on the cell-type, three different hydrogels were formed from either PEG or gelatin, which were modified with either acrylate or methacrylate moieties and then cross-linked using free radicals generated by exposure of a photoinitiator (PI) to near-UV light (Fig. 1a, blue optical path). These choices for hydrogels were dictated mainly by a model for capillary mechanics in which the interstitium provides significant mechanical support13,14,15.
Due to the high water content, PEGDA and Gel-MA hydrogels possess physical properties that resemble soft tissue. PEGDA scaffolds, in particular, are used prevalently for tissue engineering19. They are biocompatible and the porosity and mechanical properties can be easily tailored. One motivation for choosing Gel-MA was that it naturally incorporates cell-binding motifs like matrix metalloproteinase (MMP), a major factor in angiogenesis, along with RGDs and elastin, but it is soft20. On the other hand, PEGDA hydrogels are especially advantageous because they can be made stiffer and hybridized with proteolytic oligopetpides or growth factors to promote specific cell interactions, preservation and degradation19,20,21. In this work, RGDS (arginine–glycine–aspartic acid—serin) and MMP pendant groups were hybridized separately to PEGDA to enhance cell viability, promote adhesion and degradation.
Importantly, the main function of a capillary is not to perfuse blood, but rather to exchange oxygen and other nutrients for waste products between blood and the surrounding tissue. Thus, the mechanical properties—i.e. the volume, porosity and stiffness—have to be commensurate with these functions. Material penetrates through gaps between the interjoined ECs, but is impeded by the BM. The flow of blood and plasma through a capillary is driven by pressure, balanced by viscous forces. Fung et al.13 have modeled the mechanics of a capillary three ways. According to one model, if a capillary is represented by a tube floating in interstitial liquid, then the high transmural pressure is accommodated by the endothelium and the BM. Since the BM has a modulus ranging from 0.7–10 MPa, it alone could support the tension in the wall13,14. On the other hand, if the total pressure is accommodated by the whole vessel wall then, assuming it is a gel-like material, its modulus would be only about 100–400 kPa, depending on the thickness. Alternatively, a capillary can be viewed as a tunnel through a gel and the distensibility related to the ratio of the modulus of the tube wall and its surroundings13,14. Measurements of the capillary distensibility indicate that the effective modulus of the gel surrounding the tunnel would be 15 kPa14. Thus, according to this model, the interstitium could provide significant support for the capillary.
Although the bulk mechanics of hydrogels22,23 and cells24,25 have been studied separately, the nanomechanics of hydrogels incorporating cells have not. To gauge the stiffness, the elastic modulus (E) was measured by nano-indentation performed on a voxel with an atomic force microscope (AFM). The size of a voxel was determined by the volume proportion between cells and the ECM in vivo, which was supposed to be constant for each tissue26,27. The measured ratio of ECM:cells in each voxel was maintained at 1.1 ± 0.1:1. Infinitesimal strain theory relates the indentation force to the elastic modulus through the relation:  , where E denotes the elastic modulus, v the Poisson ratio and
, where E denotes the elastic modulus, v the Poisson ratio and  is a function of the indenter geometry (see Methods, supplement and Fig. S1)28. Two different probes were used: one was a blunted conical silicon tip of 7–10 nm radius (designated as the “sharp tip”, Fig. S2a left, upper inset); and the other was a tip modified by rigidly attaching a gold microsphere with a 1.5–3 μm diameter (designated as the “gold tip”, Fig. S2a left, lower inset). Regardless of the tip and the voxel size and shape, the data consistently showed a linear relationship between the indentation force and the function ϕ (Fig. S2a–c, left) as expected. However, the sharp tip consistently showed a much higher modulus compared to the gold tip (Table 1).
is a function of the indenter geometry (see Methods, supplement and Fig. S1)28. Two different probes were used: one was a blunted conical silicon tip of 7–10 nm radius (designated as the “sharp tip”, Fig. S2a left, upper inset); and the other was a tip modified by rigidly attaching a gold microsphere with a 1.5–3 μm diameter (designated as the “gold tip”, Fig. S2a left, lower inset). Regardless of the tip and the voxel size and shape, the data consistently showed a linear relationship between the indentation force and the function ϕ (Fig. S2a–c, left) as expected. However, the sharp tip consistently showed a much higher modulus compared to the gold tip (Table 1).
The disparity between the moduli obtained from the same voxel with different tips likely indicates defects in the gel network on a micrometer-scale. Micro-domains of variable stiffness like these have been observed before and attributed to so-called spatial gel inhomogeneities due to the cross-link density distribution29. According to elasticity theory, the modulus is supposed to be proportional to the absolute temperature and the cross-linking density, so that  where NA is Avogadro’s constant and kB is the Bolztmann’s constant. Thus, the data implies that the cross-linking density changes about 30-fold in PEGDA when the deformed volume changes from ~1000 nm3 to ~1 μm3. Likewise, for Gel-MA, there must be a corresponding 5-fold change in cross-linking density. Moreover, the cross-link density was about 10-fold higher in PEGDA compared to Gel-MA.
where NA is Avogadro’s constant and kB is the Bolztmann’s constant. Thus, the data implies that the cross-linking density changes about 30-fold in PEGDA when the deformed volume changes from ~1000 nm3 to ~1 μm3. Likewise, for Gel-MA, there must be a corresponding 5-fold change in cross-linking density. Moreover, the cross-link density was about 10-fold higher in PEGDA compared to Gel-MA.
Interestingly, the mechanical properties of voxels incorporating cells can reflect either the cell or the gel modulus, depending on the scale of the measurement (Table 1). The apparent elastic modulus for a voxel including an hMVEC was determined to be EPEG+cell = 110 ± 20 kPa (Fig. S2c, left) using a sharp tip, which seems to reflect the PEGDA modulus described above, not that of an EC. In contrast, indentation with a gold tip revealed a modulus of EPEG+cell = 4.0 ± 0.9 kPa, similar to other ECs measured alone24 or just PEGDA measured with a gold tip. Likewise, the modulus of a Gel-MA voxel incorporating an hNF measured with a sharp tip was EGel+cell = 35 ± 1 kPa, which was similar to that of a fibroblast25, whereas the stiffness measured with a gold tip was only 2.0 ± 0.6 kPa, which was more indicative of Gel-MA. These observations were relevant in the construction and function of a capillary in several ways. When the gel network equilibrates under a mechanical constraint imposed by the surrounding cytoarchitecture, there will be an anisotropic, inhomogeneous distribution of molecules, including water. Calculations show that, near the hNF-Gel-MA interface, the stress may be high and the concentration of water molecules will be greatly reduced within a ~1000 nm3 volume30. On the other hand, near the hMVEC-PEGDA interface, the concentration of water will not be so affected because the gel swells less when the network is more densely cross-linked.
A lower modulus implies a sparser cross-link density in the gel and higher diffusivity through it. To corroborate this assertion, the mesh size was inferred from the swelling ratio Q = Weq/Wdry obtained by weighing a voxel to determine equilibrium weight (Weq) and dry weights (Wdry)31. To achieve sub-picogram resolution, the weight was inferred from the shift in the resonant frequency of an AFM cantilever that occurs when the cantilever was loaded with a voxel. When cantilevers were loaded with PEGDA or Gel-MA, a shift in the resonant frequencies of ΔfdryPEG = −0.29 ± 0.02 kHz and ΔfwetPEG = −2.60 ± 0.05 kH and ΔfdryGEL = −6.2 ± 0.1 Hz and ΔfwetGEL = −67 ± 9 Hz were observed (Fig. S2a,b, right). The corresponding porosity,  , which was estimated to be 90.1% and 90.7% for PEGDA and Gel-MA respectively, factors into the effective diffusion coefficient through the porous medium. To verify this contention, the diffusion of a surrogate protein, fluorescent streptavidin, was also tracked through the hydrogels using confocal microscopy (see supplement and Fig. S3a,b). Thus, with these scaffolds, the modulus of the endothelium could be engineered to be locally much stiffer (>100 kPa) than the interstitium (<40 kPa) as it supposedly is in a bona fide capillary13,14 while still allowing for nutrient and protein diffusion.
, which was estimated to be 90.1% and 90.7% for PEGDA and Gel-MA respectively, factors into the effective diffusion coefficient through the porous medium. To verify this contention, the diffusion of a surrogate protein, fluorescent streptavidin, was also tracked through the hydrogels using confocal microscopy (see supplement and Fig. S3a,b). Thus, with these scaffolds, the modulus of the endothelium could be engineered to be locally much stiffer (>100 kPa) than the interstitium (<40 kPa) as it supposedly is in a bona fide capillary13,14 while still allowing for nutrient and protein diffusion.
Creating a Capillary with LCL
To determine if these mechanics could support blood flow, hMVECs, hPPs and hNFs were assembled layer-by-layer on cell-specific scaffolds into an architecture reminiscent of a human capillary in the cross-channel spanning the gap between the two microfluidic channels (Fig. 1a). Generally, the basement layer was formed from a contiguous 3×30 bed of voxels comprised of hMVECs and PEGDA-RGDS that, in turn, were flanked on either side by voxels comprised of hPPs in PEGDA-RGDS, at a ratio of 10:1 (hMVECs:hPPs) as found in vivo (Fig. 2a)32. Subsequently, the microscope focus (used for the tweezers and photopolymerization) was shifted along the optic axis 25 μm above the basement layer using a piezo-drive and then a second (interior) layer, consisting of two 1×30 columns of hMVECs with a gap 5–20 μm wide between them, was formed to create the lumen of the capillary (Fig. 2b). Again, the abluminal volume was flanked by hPPs voxels that reflect the ratio found in vivo. Similarly, a third, “attic” layer with the same constituency of cells was assembled from a 2×30 column of hMVECs voxels situated directly above the cells in the second layer (Fig. 2c). Finally, the synthetic capillary was flanked by voxels comprised of hNF cells in Gel-MA, at a ratio of 1:5 to (hNF:hMVECs) to capture the constitution of the interstitium33 as they likely play a key role in morphogenesis33,34. The total time to create a capillary this way, including loading the sample into the microfluidic, stabilizing the flow in the microfluidics, the step-and-repeat lithography, ranged from 30 to 90 min, depending mostly on the capillary length and the cell flux.
In an actual capillary, the endothelium forms the lining of the intraluminal surface and thus, the inter-endothelial contact controls the transport of solutes and circulating cells into the interstitial tissue and vice versa. The hMVECs express at least two transmembrane molecules specific for inter-endothelial contacts: PECAM-1 (platelet endothelial cell adhesion molecule-1 or CD31) and vascular endothelial cadherin (VE-cadherin). To visualize the intraluminal surfaces, the hMVECs were positively stained for CD31 (Fig. 2f–h). Likewise, the hPPs and hNFs were positively stained for NG2 (chondroitin sulfate). Reconstructed volumetric data obtained from immunofluorescence (IF) of the hMVECs, hPPs and hNFs labeled in situ showed a continuous capillary (Fig. 2f) and an unobstructed lumen with a 140 ± 43 μm2 cross-section about a quarter-millimeter-long (Fig. 2g,h), corresponding to a median diameter of 13.2 ± 2.5 μm2 that is comparable to in vivo estimates for the diameter of normal human nail-bed capillaries that range from 5 to 12 μm35. Capillaries up to a half-millimeter-long (Fig. S5a-e) were produced this way, corresponding to the average length of a human capillary, which is between 0.4 and 0.7 mm36.
The endothelial phenotype has been classified as continuous, fenestrated or sinusoidal37,38,39,40,41,42. The ECs comprising continuous capillaries form an uninterrupted lining with tight junctions between them allowing only water and ions to pass through their intercellular clefts, whereas fenestrated capillaries are more porous and allow a limited number of large molecules (proteins) to diffuse through to the interstitium and finally, sinusoidal or discontinuous capillaries exhibit large openings in the endothelium that allow blood cells and plasma to pass through. The cells comprising these synthetic capillaries generally exhibited localization of CD31 on the cell surface predominately (e.g. Fig. 2g, upper right inset). Using specific surface markers (e.g. CD31, ZO-1 and plakoglobin) and cell morphology, it was observed that cell adhesions between adjoining ECs were not tight, however. The gaps observed between adjacent hMVECs likely allow for fluid exchange between the plasma and the surrounding tissue (see the next section). Thus, the cytoarchitecture of these constructs was reminiscent of either a fenestrated or sinusoidal capillaries.
In contrast with LCL, the growth of new blood vessels by angiogenesis can be slow (>7 da)43,44,45,46,47,48,49,50,51,52,53. To accelerate vessel formation, in situ vasculogenesis has been used, which is the spontaneous formation of undifferentiated ECs into blood vessels. Alternatively, pre-vascularized tissue, formed by either angiogenesis or vasculogenesis in 3D gels49,50,51,52,53 or on patterned surfaces or by 3D printing7 can shorten the time to vascular perfusion. Finally, tube assays in which human ECs and pericytes co-assemble form tubular networks in 12 h or less17,50. However, to our knowledge, there has been no report of mature capillaries with tight junctions forming this way. Vasculogenesis results from a sequence of events mediated by cell–cell and cell–matrix interactions and flow conditions, but the mechanisms of maturation and specialization of microvasculatures have yet to be elucidated49,50. In all of these approaches, the cells, cues and micro-scale flow play key roles in morphogenesis50. It should be possible to discover the mechanisms underpinning maturation of a microvasculature by tracking remodeling in degradable hydrogels under suitable culturing conditions.
It has been hypothesized that exposure of ECs to mechanical forces associated with blood flow and growth factors will affect integrin-, Rho GTPase- and SRF-dependent signaling that control vessel remodeling and produce a mature endothelium, but it remains untested50. To test this hypothesis, systematic strategies will have to be employed involving the utilization of different cell combinations with cell-specific ECMs that promote remodeling and morphogenesis, such as PEGDA hybridized with recognition motifs such as MMP, along with different GF-supplemented media and mechanical forces to produce a mature endothelium. This can be accomplished in a synthetic capillary.
To illuminate the prospects, synthetic capillaries were created with the same methods illustrated in Fig. 2, but using a degradable PEGDA-MMP scaffold for the hMVECs instead of PEGDA-RGDS and then the time progression of the cell morphology was tracked with confocal microscopy for up to 12 h with images acquired at regular 30 min intervals with and without whole human blood flowing through them (Fig. 3a). Remodeling of the endothelium was evident within 6 h (white arrows)—in some cases, cells pressed tight against each other to form flat broader junctions while in other cases protrusions sprouted from the cells (Fig. 3a). It was also observed that the hMVECs forming the lining become polarized when subjected to shear force associated with human blood flow under differential pressures up to 1 kPa for up to 24 h, (Fig. S4). Using established protocols54, the polarization of the cells was tracked by following the position of the Golgi apparatus stained with BODIPY-FL-C5-ceramide. Confocal microscopy revealed the localization of the Golgi apparatus in hMVECs (white arrows, Fig. S4) near the lumen that constituted the 2nd layer of a synthetic capillary, indicating the onset of polarization of hMVECs. Although force is only one element driving maturity, this preliminary evidence indicates the prospects for testing hypotheses regarding remodeling and maturation of the endothelium.
As another illustration of the versatility and precision of the tool, constructs that resembled branched arterioles (in size) consisting of hMVECs and human vascular smooth muscle cells (hVSMCs) were also produced using LCL (Fig. 3b,c and S5 f-h). To visualize the intraluminal surfaces, the hMVECs were positively stained for CD31 and likewise, the hVSMCs were positively stained for CD130a (PDGFRα). Reconstructed volumetric data obtained from CD31-stained hMVECs showed a continuous bifurcated capillary (Fig. 3c) and an unobstructed lumen with a 616 ± 39 μm2 cross-section, branching into two vessels; one with a 232 ± 40 μm2 cross-section and another with a 365 ± 49 μm2 cross-section. Creating these types of structure is especially important because in vivo adhesion depends on the anatomical, geometrical and physiological fluid flow conditions as well as the interactions with the endothelium. In particular, bifurcations disturb blood flow, which predisposes them to higher adhesion relative to a straight channel55.
The flow of blood and nutrients (oxygen)
The role blood flow, perfusion and local oxygen consumption play in this construct had to be tested to prove out the model. Blood is a non-Newtonian fluid with a viscosity that depends on the volume occupied by the erythrocytes (hematocrit) and decreases with shear rate. To assess how the cytoarchitecture and mechanical properties affect the hemodynamical flow, a pressure gradient, ΔP, ranging up to 2.5 kPa was imposed across the microfluidic channel connecting to the capillary lumens with cross-sections of 56 ± 7 μm2 and 457 ± 72 μm2 and then 10% washed human erythrocytes in PBS was perfused through them (Fig. 4 and videos V1,V2,V4-V4).
A comparison of a sequence of high-resolution confocal transmission images revealed the axial erythrocyte velocity, Vax, (Fig. 4a,b) corresponding to the local cross-sectional area of the lumen (defined by fluorescent nanospheres that adhered to the adluminal surface after the erythrocytes were forced through it—Fig. 4c). Due to their volume, the erythrocytes avoided the capillary wall and so a histogram of the velocity profile measured with 2 μm resolution transverse to the capillary axis (Fig. 4d) was found to be blunter than a parabola (dotted line) indicative of a Newtonian fluid. In addition, as expected, the velocity gradient or the shear rate near the wall was steeper than that derived from a quadratic velocity profile. The peak axial velocity, which depended on the luminal area and the pressure (Fig. 4e), was comparable to that measured in vivo in capillaries and post-capillary venules of the bulbar conjunctiva in the human eye56. The wall shear stress (WSS), estimated from Newton’s law (Methods), ranged from 0.9 to about 4.5 Pa. A steep, six-fold decline (from the best fit) of WSS values as a function of the cross-sectional area was observed, which was also consistent with that found in human capillaries56,57.
Blood flow through the capillary under these conditions and the concomitant interstitial flow across it exposes the hMVECs to mechanical forces. In particular, the mechanical force exerted by blood and plasma are likely to co-direct vascular morphogenesis, which can cause vessel segments to sprout, dilate, or remain quiescent. However, the minimum shear stress required to attenuate morphogenesis in the presence of VEGF is supposed to be 0.01–0.3 Pa58. Therefore, under these conditions, endothelial sprouting was expected to be suppressed.
Oxygen is a key nutrient;59,60 its uptake ranges from 0.3–40 mol·m−3·s−1 in human tissue. Thus, a viable model also has to accommodate the transport and consumption of nutrients like oxygen. The oxygen profiles in synthetic capillaries were measured directly using the fluorescence from a medium containing a ruthenium compound (ruthenium tris-dipyridyl dichloride hexahydrate—RTDP) and a multi-core optical fiber probe61. That gradients in effectors such as oxygen develop along and transverse to the capillary axis can be appreciated from the fluorescent contours and the corresponding optical micrographs of capillaries (Fig. 5a,b, left, center). Clearly, the oxygen concentration diminished with distance along the axis of a >200 μm long capillary in Fig. 5a from the value at the inlet pO2 = 4 kPa to pO2 = 3.4 kPa at the outlet, which is consistent with uptake from viable cells in the tissue62,63. On the other hand, with a five-fold reduction in the flow, the oxygen drops from 14.0 to 7.5 kPa (Fig. 5b).
The oxygen tension along the capillary axis (Fig. 5c) can be represented by an exponential decay at a rate of Λ|| = 357 (red), 193 (green) and 393 (blue) μm depending on the inlet pressure, the differential and the corresponding flow rate. Independent of the flow rate, a gradient also develops in the interstitial pO2 as well, as the transverse distance from the capillary axis increases with an exponential decay rate of Λ⊥ = 61 ± 10 μm although it depends critically on the cell density and constituency in the interstices. Nearly anoxic conditions were observed in this tissue construct after 1 hr without flow (Figs. S6,S7), which is nevertheless consistent with the oxygen profile measured in explants62.
These data, especially the exponential decay, indicate that the oxygen supply within this engineered tissue was determined by diffusion, advection and consumption. The diffusion coefficient of oxygen in water is Dwater = 2,100 μm2·s−1, whereas the diffusivity in tissue is typically less, ranging about Dtissue = 1,750 μm2·s−1 63. Thus, the luminal wall likely reduces the diffusion of oxygen so that the measured transverse gradient reflects, not just consumption, but the cell density as well. Finite element simulations (FES) that account for the diffusivity and consumption of oxygen show that the mass transport of oxygen-rich medium through and around a synthetic capillary was sufficient to maintain physiology (Fig. 5a–d), whereas the absence of flow reduced dramatically the abluminal oxygen supply (Figs S6 and S7). The oxygen gradient was evident from the low oxygen partial pressure (blue) outside relative to the lumen of the capillary and corresponds closely with the measurements (Fig. 5a–d). The diffusivity of nutrients such as oxygen in these constructs seems to be limited by the scaffold—likely the clefts between the hMVECs—not the endothelium. Thus, these tubular micro-channels, constructed from human cells function, much like a fenestrated capillary; they successfully flow human blood (and plasma) and nutrients such as oxygen, but the oxygen diffuses through the endothelial lining.
Trapping a cancer cell in a synthetic capillary
Taken altogether, these observations support the notion that LCL could be used to engineer tissue with a microvasculature. But how would the tissue be used? To concretely illustrate the advantages and expose some of the shortcomings, synthetic capillaries were created as a naive model to explore the interactions between a breast cancer cell and the endothelium. When it is finally developed, a model like that might be used to study the first steps in extravasation that occur during metastasis. Metastases are formed by cancer cells that break away from the primary tumor, travel via the circulatory system and become lodged in capillaries at a secondary site64,65,66,67,68,69. Once lodged in a capillary, cancer cells escape or extravasate to the surrounding tissue. The first two steps in the extravasation of cancer cells through a capillary endothelium into the organ are rolling and adhesion of the cancer cell in the capillary64.
LCL was used to create capillaries with lumens graded in cross-section to trap metastatic MDA-MB-231 human breast cancer cells (Fig. 6). Subsequently, optical tweezers were used to introduce a single cancer cell into them and then EC and cancer cell growth media (1:1) were flowed through the microfluidic inlet (0.5 μL/min) to trap the cancer cell in the capillary without forcing it through. The time progression of the cancer cell in the capillary was followed with confocal microscopy for up to 48 h with images acquired at regular 10 min intervals. The progression revealed several interesting features. In eight separate observations, the cancer cell stayed dormant (data not shown). However, a few of the cancer cells were motile against the medium flow, adhering and changing morphology when making contact with neighboring hMVECs (Fig. 6 and the corresponding video V3 in the supplement) without compromising viability.
These observations of rolling and adhesion of a cancer cell in a synthetic capillary are too limited to be conclusive, but the exercise revealed aspects of the model that need development. On the one hand, real-time in vivo imaging of extravasation has been accomplished in small animals65, which can capture some facets of the complexity of human responses, but fails to capture others that are species-specific1. Likewise, it is challenging to develop high-throughput systems to evaluate hundreds of animals. In particular, an accurate model for extravasation should include whole human blood, leukocyte and platelets, since they have been implicated in adhesion64.
Among the advantages LCL offered was precise control of the cytoarchitecture and cell placement and cell-specific ECMs. The constructs used for these tests each consisted of heterogeneous, hierarchically organized populations of <100 human cells that were assembled layer-by-layer in <30 min and remained viable for >48 h. In particular, the cross-sectional area of the lumen, which ranged from 352 ± 102 μm2 to 669 ± 142 μm2 (Fig. 6) was designed and implemented so that part of the capillary would be just smaller than a typical MDA-MB-231 cancer cell, which was gauged to have an area of 460 ± 190 μm2 (n = 20 cells). Moreover, the same cytoarchitecture was reproduced repeatedly—eight other capillaries were created with practically identical cross-sections. Finally, whole human blood can be perfused through synthetic capillaries constructed from human tissue, but clotting becomes problematic without an anticoagulant (Video V4).
Even though the number of cells in these constructs was comparable to that comprising an MFU, the superstructure of an organ consists of 106-107 MFUs. Therefore, the throughput associated with LCL implemented this way was inadequate to the task of creating whole organs. However, the technology required to improve on the time-to-product from 30 min has already been established. Improvements can be made in the microfluidic conveyance to the assembly area to increase: 1. the flux, while avoiding cell aggregation; and 2. the number of optical tweezers used to manipulate cells. Mainly, it requires multiple optical tweezers controlled independently. This requirement has already been satisfied. The number of tweezers can be increased 100-fold—more than 400 cells have been trapped simultaneously70. The number is limited by the irradiance and damage threshold of the optics and AODs. Thus, hundreds of cells could be manipulated into position concomitantly under computer-control using image recognition software to acquire cells conveyed to an assembly area using laminar flow in a microfluidic channel. It is only necessary to avoid excessive shear on the cells.
In contrast with a synthetic capillary, other in vitro methods that test the ability of cells to migrate on or through a solid substratum such as scratch assays, trans-well migration assays and invasion assays, do not recapitulate the anchorage independence required for metastatic dissemination through the circulation68. Moreover, invasion assays use only a single type of ECM, which fails to account for the differences in the ECM composition of the primary and secondary sites68,69. Therefore, an in vitro model consisting of human cells in the proper social context with cell-specific scaffold is needed to properly analyze the steps in extravasation and early metastasis. Synthetic capillaries could play this role, but the constructs produced so far have been immature without tight junctions.
Conclusion
In summary, it has been established that LCL can be used to precisely control the type and position of cells on a composite, cell-specific hydrogel scaffold to create microstructures that functionally resemble a fenestrated or sinusoidal blood capillary with a nanostructure consistent with the elastic modulus and porosity of human connective tissue. In particular, with these scaffold, the modulus of the endothelium was engineered to be locally much stiffer (>100 kPa) than the interstitium (<40 kPa) as it supposedly is in a bona fide capillary13,14, while still allowing for nutrient and protein diffusion. These constructs supported the forces associated with human blood flow and nutrient gradients similar to those measured in a bona fide human capillary.
The application of LCL to tissue engineering is compelling for several reasons. First, it has a facility for precisely and repeatedly placing human cells in a specific cytoarchitecture. A capillary was chosen for illustration because it forms the basis for many MFUs, but in principle any tissue architecture consisting of a heterogeneous population of 100–1000 cells could be organized hierarchically this way to express a predictable function. With model microvasculatures such as a bifurcated arteriole (Fig. 3b,c) or an under-sized capillaries (Fig. 6), cell adhesion, which is supposed to be highly dependent on the anatomical, geometrical and physiological fluid flow conditions, can be tested. Models like these, which can be repeatedly produced, could be important for drug discovery. Second, the precision of the cell placement is submicron. This is especially important in the construction of a capillary because the lumen is so small (3–30 μm in diameter). So far, the literature has mainly focused on larger diameter (arteriole-sized) vessels that were self-assembled without the prospect for larger scale hierarchical organization. Third, not only can cell placement be controlled with precision, but so can the ECM. With LCL it is possible to tailor the microenvironment of each cell-type by photo-polymerizing a specific pre-polymer in each voxel to form cell-specific scaffolds (Fig. 2). Thus, the volume, stiffness, porosity, adhesion and degradation in the ECM can be controlled with single cell precision. Finally, a complex hierarchically-organized cytoarchitecture can be assembled and perfused in as little as 30 min, which contrasts with angiogenesis that can take a week before structures can be perfused. No other method for tissue engineering offers these features.
Materials and Methods
Cell Culture
All the cells were cultured and harvested similarly.
Human microvascular endothelial cells (hMVECs)
The hMVECs (CC-2543, Lonza) were cultured in endothelial growth medium (EGM, Lonza), according to the manufacturer’s (Lonza) specifications. Briefly, the hMVECs were maintained at 37 °C, in a humidified incubator at 5% CO2/ 95% air until they reached 70–80% confluency (6–8 da after seeding). Only cells at passage numbers 4–7 were used in this work. These confluent hMVECs reproduced the growth-arrested, quiescent state of normal blood vessels in vivo, expressing vascular endothelial (VE)-cadherin at cell–cell junctions. Subsequently, the medium was aspirated, the adherent hMVECs were then washed with 30 mM HEPES-BSS buffer (CC-5024, Lonza) and trypsinized by adding a volume of trypsin-EDTA solution (CC-5012, Lonza) sufficient (2.0 mL) to cover the cell surface in the T25 flask and then incubated at room temperature for 2–4 (5) min. Once they appeared rounded and began to detach, the cells were harvested by sharply rapping the culture flask. The reaction was stopped by adding (4 mL) of trypsin-neutralizing agent (CC-5002, Lonza). The detached cells were transferred to a sterile 15 mL tube and centrifuged at an RCF of 200×g for 5 min to pellet the cells. The supernatant was aspirated and the cell pellet was re-suspended in PBS by pipetting gently no more than 2-3 times.
Human neonatal fibroblasts (hNF)
Likewise, the hNFs (CC-2509, Lonza) were cultured in fibroblast basal medium (CC-3131, Lonza), according to the manufacturer’s specifications until they reached 90% confluence (5-7 da after seeding). After two passages, the cells were harvested essentially as described above.
Human pericytes from placenta (hPP)
Finally, hPPs (C-12980, Promocell) were cultured in growth medium (C-28040, Promocell), according to the manufacturer’s specifications until they reached 70–90% confluence (6–8 da after seeding) and then harvested as described above.
Human vascular smooth muscle cells (hVSMC)
The hVSMCs (CC-2581, Lonza) were cultured in smooth muscle growth medium, according to the manufacturer’s (Lonza) specifications. Briefly, the cells were maintained at 37 °C, in a humidified incubator at 5% CO2/ 95% air until they reached 70–80% confluency (6–8 da after seeding) and then harvested as described above.
Microfluidic Device
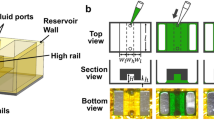
A multi-channel microfluidic device was used to convey cells to an assembly area. The microfluidic was a four-port, two-channel device with a cross-bar connection between the channels. (Fig. 1(a), lower right inset). The two entry-channels, which were 200 μm wide and 150 μm deep, merged into a single 1.2 mm-long and 600 μm-wide cross-bar laced with four-125×125 μm2 pedestals at regular intervals, which were used for mechanical support during assembly. The microfluidic device was formed from poly (dimethylsiloxane) (PDMS) using a mold-casting technique. A master mold of the design was generated by stereo-lithography (FineLine Prototyping, Raleigh, NC) and made of DSM Somos ProtoTherm 12120, a strong, high temperature tolerant plastic. The PDMS silicone polymer used to create the chips was commercially available as Sylgard 184 (Dow Corning), a two-part polymer mix. The two parts were mixed thoroughly at a 1:10 ratio of curing agent to epoxy. The mixture was degassed in house vacuum for 30 min and then poured into the master mold, where it was cured at 75 °C for at least 15 h. After cooling to room temperature, the plastic, which hardened to a rubber-like consistency, was peeled away from the mold yielding the inverse pattern of the master. The microfluidic channels were connected to external pressure and fluid reservoirs through a 2 mm diameter hole at the inlet and outlet ports incorporated in the form.
Though PDMS is transparent, our microfluidic chip was thick (>4.0 mm) and light scattering through it prevented optical access through the top. Accordingly, for optical access, the bottom of the PDMS was sealed with #0, 24×60 mm (Gold Seal) cover glass. To tightly bind the PDMS microfluidic to the cover glass, a covalent bond between the PDMS and glass was formed using an oxygen plasma in a Harrick plasma cleaner (PDS-32G). The oxygen plasma generates silanol (Si-OH) groups on the surface of PDMS, which reacted with silanol groups on the glass surface to form an Si-O-Si bond. The plasma was monitored to ensure a bright, purplish color and left on for 180 sec. The PDMS chip was gripped by the sides and placed in contact with the coverslip and then a uniform pressure was applied (magnetically) at 75 °C overnight to form the bond.
To enhance hydrogel adhesion the internal surfaces of the microfluidic device were treated with a methacrylate silane treatment that crosslinked with the hydrogel. A 2% (v/v) solution of 3-(trimethoxysilyl) propyl methacrylate was made in 10 mL of 95% ethanol. The solution was adjusted to a pH of 5 using 50 μL of glacial acetic acid. 500 μL of this solution was pushed through the microfluidic chip using a 1 mL syringe and incubated for 5 min at room temperature. The chip was then flushed with ethanol (5 ml) and deionized water (5 ml) and baked in the vacuum oven at 75 °C for 30 min.
Hydrogel and Photopolymerization
The cells were partially encapsulated in hydrogels formed from either PEG or gelatin precursors that were modified with either acrylate or methacrylate moieties and crosslinked using free radicals generated by exposure of a photoinitiator (PI), 2-hydroxy-[4-(hydroxyethoxy)]-2methyl-1propanone (Irgacure 2959, Sigma) at 0.3% (w/v) to near-UV light. The hydrogel used to form the endothelium (PEGDA-RGDS) was comprised of PEG diacrylate (PEGDA) and acryl-PEG-RGDS with adhesion ligand RGDS (arginine–glycine–aspartic acid—serin, N-terminus to C-terminus, American Peptide Company) to improve the cell attachment. The peptide sequence (1 mg/ml) was covalently coupled to a mono-acrylated polyethylene glycol of molecular weight 3.4 kDa containing an N-hydroxysuccinimidyl group on one end (Acryl–PEG 3400–NHS, Laysan Bio) by reacting equimolar amounts of Acryl–PEG3400–NHS and RGDS in 50 mM sodium carbonate buffer at pH 8.2 for 2 h at room temperature. Once the reaction was complete, the product was dialyzed against water overnight using dialysis tubing of molecular weight cutoff 1.0 kDa. The dialyzed product was then lyophilized to obtain the monoacrylated PEG with a RGDS sequence (Acryl–PEG 3400–RGDS). Just prior to injection into the microfluidic, cell suspensions were combined 1:1 with a pre-polymer mixture made of 5.0 kDa polyethylene glycol diacrylate (PEGDA, Laysan Bio) at 10% (w/v) in the final solution, 3.4 kDa PEG-RGDS at 0.2 mM; cell media; and PI.
Following other work21, to promote remodeling of the endothelium, a degradable PEGDA-MMP was also synthesized using MMP-sensitive peptides by reacting the peptide chain GCRDVPMSMRGGDRCG (GenScript, Piscataway Township, NJ) with the succinimidyl valerate ester (SVA) on an acryl-PEG2000-SVA (Laysan Bio Inc., Arab, AL) in 50 mM sodium bicarbonate (pH 8) at a 1:2 mole ratio for 4 h. The reaction mixture was then dialyzed for 24 h to remove unreacted reagents, lyophilized and stored at −80 °C until use.
Finally, methacrylated gelatin (Gel-MA) was used to encapsulate fibroblast on the abluminal surface of the capillary. The Gel-MA was synthesized from porcine skin. Type A porcine gelatin was mixed at 10% (w/v) into Dulbecco’s phosphate buffered saline (DPBS) at 60 °C and stirred until fully dissolved. Methacrylate anhydride (Sigma) was added to the methacrylate 50% of the lysine group at a rate of 0.5 mL/min to the gelatin solution under stirred conditions at 50 °C. This mixture was reacted for 1 h. The fraction of the lysine groups reacted was modified by varying the amount of methacrylate anhydride present in the initial reaction mixture. Following a five-fold dilution with additional warm (40 °C) DPBS to stop the reaction, the mixture was dialyzed against distilled water using 12–14 kDa cutoff dialysis tubing for 1 week at 40 °C to remove salts and methacrylic acid71. The solution was then lyophilized for 1 week to generate white porous foam and stored at −80 °C until it was used. Just prior to injection into the microfluidic, cell suspensions were combined 1:1 with the Gel-MA precursor 5–20% (w/v); in PBS; and a photoinitiator, 2-hydroxy-[4-(hydroxyethoxy)]-2methyl-1propanone at 0.5% (w/v). (Irgacure 2959, Sigma).
All the cell-precursor suspensions were mixed well to achieve a homogeneous mixture by slowly pipetting 3–4× and then injected into the microfluidic using a syringe pump through a 23G-gauge needle. The hydrogel was photo-polymerized in the microfluidic using near-UV light from a metal halide light source (X-CITE 120Q, Lumen Dynamics) that was bandpass filtered (λ = 360 ± 12 nm) UV filter (Semrock) at an irradiance of 5.0 ± 0.8 W/cm2. The pre-polymer-cell suspension was exposed to the near-UV light for 400 ms. A rectangular mask was placed in the UV path to control the size and shape (15×15 μm2) of the hydrogel at the objective focus. Immediately after each microarray was formed, the cell array was flushed with fresh pre-polymer mixture. At the end of the lithography, the microfludic was flushed with fresh culture medium.
Live Cell Lithography
Cells in the assembly area in the cross-bar of the microfluidic device were organized into 3D, heterogenous arrays using multiple optical tweezers. The tweezers were formed using a Zeiss Fluar 40× oil immersion objective (1.3 NA) in a Zeiss Axio Observer inverted microscope with light from a tunable, CW Ti:sapphire laser (3900S, Spectra Physics) operating at λ = 830 nm, pumped at 532 nm by a 15 W Nd:YVO4 diode-pumped solid state laser (Millenia Pro15J, Spectra Physics) as described elsewhere11,18,70. To create multiple, time-multiplexed tweezers, a single laser beam was deflected transverse to the direction of propagation using two orthogonally mounted acousto-optic deflectors (AOD, AA-Optoelectronic), optimized for maximum diffraction efficiency at the wavelength of interest (860 nm), to give independent control of the x- and y-positions of a beam, allowing for the creation of a two-dimensional (2D) array of traps. The AODs were used to create 2D, 3×3 arrays of time-multiplexed optical tweezers that were used to pluck cells from a laminar flow and steer them into the array. The time-averaged power in each trap within the array was typically <8 mW. Laser power was recorded as the time-averaged power based on the duty cycle (i.e. the number of traps) in the time-shared array.
The laser beam was scanned rapidly from one trap to the next in the 2D array, dwelling at the desired trap position just long enough to illuminate it and fix the location of a cell there. When the AOD deflected the beam to the next trap location in the array, cells that were not illuminated diffused from the target locations and were dispersed. To prevent dispersal, the rate of deflection between traps was carefully chosen relative to the time the cell spent in the dark. Typically, the light/dark time for each cell was 20 μs/ 100 μs. The cellular arrays were not limited to planar configurations; the optical tweezers could be offset along the optic axis using a computer-controlled piezo-stage upon which the microfluidic device rested as an elevator. Once it was in position, a cell was partially encapsulated in hydrogel. Handling one cell every 5–20 sec was typical. Following this approach, capillaries were assembled voxel-by-voxel, layer-by-layer. When a capillary was completed, the cell suspension was flushed and a fresh volume was introduced into the microfluidic to avoid prolonged exposure to free radicals.
The microfluidic device, along with the entire Zeiss Axio Observer inverted microscope, was encased in an environmental chamber (XL S1, Pecon/Zeiss, Switzerland). After assembly, to maintain the tissue, the chamber was used to automatically regulate using feedback: the temperature (37 ± 0.5 °C), the partial pressure of CO2 (pCO2 5% atm). To establish the optimal wavelength and power for tweezing cells, viability was tested by partially encapsulating them in hydrogel (along with dark controls) and assaying them 10 h later with green-fluorescent calcein-AM, which indicated intracellular esterase activity and red-fluorescent ethidium homodimer-1 that assessed the plasma membrane integrity (Molecular Probes, C3100MP).
Testing the Mechanical Properties of the Hydrogel with AFM
To test the modulus, nano-indentation tests were conducted with an MFP-3D-BIO AFM (Asylum/Oxford; Santa Barbara, CA) interfaced to an inverted optical microscope (Axio-Observer Z1, Zeiss). The AFM employed a low noise Z-sensor coupled with an ultra-quiet Z-drive to produce noise in the tip-sample distance <30 pm at 1 kHz bandwidth. To minimize drift and reduce acoustic noise, the inverted optical microscope was mounted on an optical air table with active piezoelectric vibration control (Stacis, TMC, Peabody, MA), housed in an acoustically isolated, NC-25 (Noise criterion) rated room in which the temperature was stabilized to less than ± 0.1 °C over 24 h through radiative cooling. The Z-piezo sensor (Z-sensor) was calibrated using a standard calibration grating (NT-MDT, Moscow, Russia). The deflection sensitivity was calibrated by pressing the tip against a freshly cleaved mica surface and correlating the cantilever deflection to the Z-sensor reading. With these precautions, detector noise is <0.1 nm/√Hz for frequencies above 0.1 Hz.
An AFM cantilever was used to deform a small volume in a voxel. The voxels were created on a #1 cover-glass both with and without cells, immersed within an area defined by a silicone isolator (Grace Bio-Labs) filled with PBS at 37 °C. Typically, voxels were formed with a volume of 50×30×30 μm3. Prior to polymerization, the cover glass was solvent-cleaned with acetone, isopropyl alcohol and 18 MΩ de-ionized water and then conditioned with a 20% oxygen plasma at 25 W (Harrick Plasma) for 3 min. Whereas PEGDA voxels require no surface treatment to adhere, the coverslip used to support the Gel-MA voxels was coated with 3-(trimethoxysilyl) propylmethacrylate.
Two types of cantilevers were used to deform the voxels: one with a blunted conical silicon tip of 10 nm radius (Fig. S2a left, upper inset) with a cone angle of about 2α = 56° (PPP-ContSC, Nanosensors); and another with a tip (CP-PNP-Au, NanoandMore) modified by rigidly attaching a gold microsphere with a 2 μm diameter (Fig. S2a left, lower inset). The spring constants, k, typically, 130–260 pN/nm for the sharp tip and 17–43 pN/nm for the gold tip, were determined by measuring the thermal noise spectra and fitting the response to a simple harmonic oscillator. The resonant frequency, f0 and quality factor were obtained by measuring the thermal noise spectra of the unloaded AFM cantilever in liquid (PBS) and fitting the response to a simple harmonic oscillator with an added noise floor: i.e.
 where Λ represents the quality factor. The cantilever sensitivities were determined consistently to be near 50 nm/V.
where Λ represents the quality factor. The cantilever sensitivities were determined consistently to be near 50 nm/V.
The cantilever was positioned directly above a voxel using a 40× telescope and then force-extension curves were measured using a low rate (200–1000 nm/s) z-scan at a frequency <1Hz with a range as large as 1 μm, which minimized not only hysteresis but also drag force. Five replicate indentations in the same location were done to establish that the material properties of the voxel did not change with repeated indentations. Finally, the indentation force (F) was calculated from Hooke’s law (F = k·d), where k and d denote the cantilever’s spring constant and the cantilever’s measured deflection, respectively. The indentation depth (δ) was calculated from the difference in the z-sensor and the deflection of the cantilever. To minimize mechanical strain of the underlying voxel and to ensure an exclusive indentation of the top of the cell layer only, the maximum applied force was limited to 400–2000 pN.
The indentation force was related to the elastic modulus through the relation:  where E denotes the apparent elastic modulus and v denotes the Poisson ratio28. The function
where E denotes the apparent elastic modulus and v denotes the Poisson ratio28. The function  corresponds to a rigid sphere of radius R indenting an elastic half-space to a depth δ—an adequate model for a microparticle-modified tip used in some of the tests. On the other hand, when an unmodified AFM tip was used, it was represented by a blunt cone with tip angle of 2α, so that:
corresponds to a rigid sphere of radius R indenting an elastic half-space to a depth δ—an adequate model for a microparticle-modified tip used in some of the tests. On the other hand, when an unmodified AFM tip was used, it was represented by a blunt cone with tip angle of 2α, so that:

where a denotes the radius of contact, b is the transition radius, R the tip radius and α is half the cone angle of the tip28.
Weighing Hydrogel with an AFM
Voxels without cells were analyzed for swelling ratio, porosity, average molecular weight between crosslinks (Mc, g/mol) and mesh size (ξ) by depositing them on an AFM cantilever and weighing them using the change in the resonance frequency. The swelling ratio Q and water content were computed from wet weight (Wwet) and dry weight (Wdry) measurements. Samples were weighed wet, lyophilized for days and then weighed dry. Two types of cantilevers were used to weigh the voxels: one was a V-shaped, gold-coated nitride probe (BL-TR400PB, Olympus, Japan) with a nominal spring constant of 90 pN/nm (Fig. S2a right, inset), which was used in conjunction with iDrive (Asylum), for easy auto-tuning of the cantilever in fluid; and the other (Fig. S2b right, inset) was a rectangular, silicon single beam cantilever (SSS-FM, Nanosensors) with a nominal spring constant of 2.8 nN/nm.
To weigh the hydrogels, the cantilever was first cleaned in solvents and then the resonance frequency was stabilized after repeated rinses in 18 MΩ de-ionized water. The resonance frequencies, 32.45 kHz and 72.05 kHz for V-shaped and rectangular cantilevers respectively, were each measured 10 times using a sweep width of 10 kHz and drive amplitude of 50 mV and then averaged for the calculation of the loaded mass. Then, a hydrogel was formed on the cantilever (Fig. S2a,b, lower right insets), which resulted in a reduction in the resonance frequency. With the load distributed uniformly along the cantilever, the change in the resonant frequency is proportional to the change in mass (Δm) through the relation:  in which the resonance frequency is
in which the resonance frequency is where m denotes the mass and k the spring constant. The resonant frequency was obtained by measuring the thermal noise spectra of the unloaded AFM cantilever in air and fitting the response to a simple harmonic oscillator. For the V-shaped cantilever, with a resonant frequency of
where m denotes the mass and k the spring constant. The resonant frequency was obtained by measuring the thermal noise spectra of the unloaded AFM cantilever in air and fitting the response to a simple harmonic oscillator. For the V-shaped cantilever, with a resonant frequency of and a mass of 12.4 ng, the mass resolution was about
and a mass of 12.4 ng, the mass resolution was about  . Likewise, for the rectangular cantilever, with
. Likewise, for the rectangular cantilever, with  and
and  ,
,  .
.
To accommodate the non-uniform loading of the cantilever associated with the hydrogel the spring constant was modified according to:  , where L is the length of the cantilever and ΔL is the distance from the cantilever center-of-mass of the voxel31. This accommodation applies for both V-shape and rectangular cantilevers. For the V-shape cantilever weighing PEGDA, L = 100 μm and ΔL = 35 μm, whereas for the rectangular cantilever weighing Gel-MA, L = 225 μm and ΔL = 60 μm. Thus, the non-uniform load on the cantilever was determined from the frequency shift (Table S1). After the wet weight was measured, the cantilever with a wet equilibrium hydrogel was transferred to the lyophilizer and freeze-dried under conditions of 4 Pa and −87 °C until no further change was observed (4–9 da) and then the dry weight was measured. The swelling ratio
, where L is the length of the cantilever and ΔL is the distance from the cantilever center-of-mass of the voxel31. This accommodation applies for both V-shape and rectangular cantilevers. For the V-shape cantilever weighing PEGDA, L = 100 μm and ΔL = 35 μm, whereas for the rectangular cantilever weighing Gel-MA, L = 225 μm and ΔL = 60 μm. Thus, the non-uniform load on the cantilever was determined from the frequency shift (Table S1). After the wet weight was measured, the cantilever with a wet equilibrium hydrogel was transferred to the lyophilizer and freeze-dried under conditions of 4 Pa and −87 °C until no further change was observed (4–9 da) and then the dry weight was measured. The swelling ratio  and the porosity (Q-1)/Q were inferred from these weights.
and the porosity (Q-1)/Q were inferred from these weights.
Estimating the Mesh Size, ξ
The molecular weight between crosslinks can be approximated using the Peppas-Merrill model according to:72

where Mn is the average molecular weight of the polymer, v is the specific volume of bulk polymer in the amorphous state, V1 is the molar volume of the solvent, μ is the Flory-Huggins polymer-solvent interaction parameter and V2,r and V2,s are the volume fraction of the polymer at the relaxed state and swelling equilibrium, respectively. V2,r is equivalent to the volume concentration of the solution where cross-linking occurs and V2,s = 1/Q. Accordingly, the mesh size (ξ) was calculated from:  where l is the average value of the bond length between C-C and C-O bonds in the polymer repeat unit, Mr is the molecular mass of the repeat unit [-O-CH2-CH2-] and Cn = 4 is the characteristic ratio for polymer. Similarly, for Gel-MA, Mc was estimated from the Peppas-Merrill model, but with V2,r = 1. In this case, the mesh size was given by:
where l is the average value of the bond length between C-C and C-O bonds in the polymer repeat unit, Mr is the molecular mass of the repeat unit [-O-CH2-CH2-] and Cn = 4 is the characteristic ratio for polymer. Similarly, for Gel-MA, Mc was estimated from the Peppas-Merrill model, but with V2,r = 1. In this case, the mesh size was given by:  where l is the mean of one C-C bond and two C-N bonds (so that the factor 2 was replaced by 3). The parameters characterizing each hydrogel are tabulated in Table S2.
where l is the mean of one C-C bond and two C-N bonds (so that the factor 2 was replaced by 3). The parameters characterizing each hydrogel are tabulated in Table S2.
Microscopic characterization of synthetic capillaries
Cross-section of a capillary
A 3D model for each microtubule was obtained from the volume reconstructed from image stacks obtained from a series of confocal images of the cellular array (Imaris, AG Switzerland). Carboxylate-modified, mono-dispersed fluorescent nanospheres (40 nm diameter, FluoSpheres) were perfused through the capillary and attached to cell walls (Molecular Probes, Invitrogen). Generally, the cross-section of the lumen transverse to the capillary axis was irregular, so the area, not the diameter, was measured at 5 μm intervals. The coincidence between these areas and velocities along the capillary axis were obtained for several capillaries, varying the differential pressure across the capillary and average area of the lumen.
Velocity and Shear Stress
To flow blood through a capillary, first it was tightly sealed against the pedestals and walls forming the cross-connecting channel in the microfluidic device using 5 kDa PEGDA hydrogel. Then a washed 10% suspension of human erythrocytes in PBS containing sodium citrate was perfused through the capillary using a microfluidic control system (MFCS, Fluigent, Paris, France) to determine the pressure and monitor the flow through it. Focusing on the lumen along the axis of the capillary (x-y), time-sequenced data of the trajectories associated with individual erythrocytes was extracted from confocal scanned transmission images. Each frame was subtracted from the previous one to generate a video of the pixels that have changed (Fiji software). After this operation, only the outline of the erythrocytes remained in the image sequence.
These images were subsequently digitally filtered to eliminate noise. The trajectory of each erythrocyte was tracked throughout the capillary by identifying the x and y coordinates corresponding to the center of the cell in each consecutive frame using the MTrackJ plugin. The axial erythrocyte velocity Vax was measured using the definition: Vax = Dax/Δt where, Dax denotes the displacement of an erythrocyte between frames, after a short time interval Δt, which corresponds to the frame acquisition rate (8–32 ms). The erythrocyte velocity was averaged and a standard deviation was calculated. For erythrocytes moving slowly, more than 2Δt time increments were used for each Vax measurement, so that the minimum distance traveled was about 5 μm. The maximum relative error in the axial velocity measurement can be regarded as the combined effect of the relative error in the measurement of the time interval Δt between two successive images of the flow and the relative error in the measurement of the axial displacement of an erythrocyte, but since the former was negligible (deviations of the frame acquisition rates were less than 0.001%), the total relative error in the axial velocity measurement was equal to the latter, i.e. approximately 5%, which was attributed to the optical resolution of the image (500 nm/pixel). However, whereas the measurement was accurate the actual movements of the RBCs were random as evident from the error bars representing the standard deviation in position.
For erythrocytes flowing in a capillary, there was a net hydrodynamic force pushing the cells towards the center. Erythrocytes did not penetrate the capillary wall, which implies that they must be at least ½ dRBC away, where dRBC is the diameter of a human erythrocyte. This means that, on average, there will be more erythrocytes near the center of the capillary. The average axial velocity, Vax and the wall shear rate were measured directly. The average wall shear rate always exceeded 200 s−1, which makes the fluid appear Newtonian. In addition, the in vivo viscosity law was used for the estimation of η to take into account the viscosity change with vessel diameter. The wall shear stress was estimated from Newton’s law, which is just a product of the gradient of the velocity profile with respect to the outward normal n of the wall and the kinetic viscosity, η, i.e.  , assuming the viscosity of blood for shear rates >100 s−1 at 37 °C was given by η = 3.5 mPa·s56,57. Typically, the shear rate in a capillary ranged up to about
, assuming the viscosity of blood for shear rates >100 s−1 at 37 °C was given by η = 3.5 mPa·s56,57. Typically, the shear rate in a capillary ranged up to about  .
.
Fluorescence Imaging
Cells
Fluorescence data was collected using a Leica TCS SP5 II (Leica Microsystems) confocal microscope with an 8 kHz resonant scanner and enhanced, hybrid GaAsP (HyD) detectors for improved sensitivity to fluorescence. All confocal images were acquired using either a 63× 1.2 NA or 25× 0.9 NA water immersion objective (Leica) with an argon laser excitation (488 or 514 nm) at 500 nW and diode laser at 564 nm using a pinhole (1 Airy unit). Fluorescent z-stacks (along the optic axis) were recorded. An image matrix comprised 512×512-pixels, each was 0.4 μm thick extending over a 150.0 μm depth. False-color perspective volumes were reconstructed from image stacks obtained from a series of confocal images showing the 3D aspects of the cellular array using Imaris software (Bitplane AG), which rendered fluorescent volumes using an intensity threshold.
The entire confocal microscope including the microscope stage, objective and the microfluidic device, were encased in an environmental chamber (BLX 2000). To maintain the tissue during incubation, the chamber was used to automatically regulate using feedback the temperature (37 ± 0.1 °C) and the partial pressure of CO2 (pCO2 5% atm).
Immunofluorescence (IF) Labeling of Cells
To visualize cell-specific surface markers, the cells comprising the capillary were stained by direct IF using FITC-conjugated mouse anti-human CD31 monoclonal antibody (303103, Biolegend, San Diego, CA) and CF680-conjugated mouse anti-human chondroitin sulfate monoclonal antibody (554275, BD Bioscience, San Jose, CA). Prior to direct IF, the anti-chondroitin sulfate antibody was conjugated with CF 680 (Biotium, Inc., Hayward, CA). To validate cell-cell adhesion in confluent hMVECs maintained under standard culture conditions, FITC-conjugated mouse anti-human VE-cadherin monoclonal antibody (NBP1-58370, Novus Biologicals, Littleton, CO) was used. For live cell staining, these antibodies were diluted using culture medium and used at the final concentration of 1-2 μg/ml. After incubation at 37 °C, in 5% CO2 for 30 min., unbound free antibody was washed with excess culture medium. Attempts were made to label hNFs with CD-90 (Thy1) without marking the pericytes or hMVECs, but contrary to expectation, Thy1 failed to stain hNFs.
Oxygen
To rigorously establish the boundary conditions and eliminate spurious oxygen, the microfluidic device was housed in an environmental chamber (Pecon GmbH, Erbach, Germany) that was purged with nitrogen for >1 h for a low oxygen condition or backfilled with oxygen for high level conditions. To preclude out-diffusion of oxygen, the connections to the microfluidic were made with Polyetheretherketone (PEEK) tubing. After a capillary was formed, oxygenated fluid was forced through it using a differential pressure across the inlets; the right-hand port on the inlet of the microfluidic (Fig. 1a) was connected to a high-level oxygenated medium, pO2 = 20 kPa, flowing at a 30 μL/min rate, while low-level oxygenated medium, pO2 ≤ 2 kPa, flowed at a 0.5 μL/min rate into the left-hand inlet.
The pressure within the microfluidic device was estimated from a finite element analysis using COMSOL. The Navier-Stokes equation was solved, taking into account the tubing from an ELVE control system (Elveflow, Paris, France) to the microfluidic and from the microfluidic to the waste containers and the applied flow rates. It revealed an average flow rate of 0.25 and 0.05 μL/min, corresponding to differential pressures of 1.2 Pa and 0.045 Pa across the capillaries in Fig. 5a,b respectively. The low-level oxygenated solution was prepared by pushing degassed medium purged with dry nitrogen gas through Tygon tubing (ID 500 μm) 1 m long at a flow rate of 50 μl/min, whereas the high-level solution was prepared by pushing medium with atmospheric oxygen through the same Tygon tubing 1 m long at the same flow rate. The solutions were subsequently forced through PEEK tubing to prevent further out-diffusion.
To detect oxygen, a ruthenium dye RTDP (tris(2,2-bipyridyl)dichloro-ruthenium(II) hexahydrate) (0.3% w/v) was used in conjunction with the NeoFOX phase measurement system (Ocean Optics). The excitation light of the pulsed blue LED and a red reference LED with a repetition rate of 94 kHz were focused with a 100× objective onto a ~20 μm spot on the sample and on the lumen of the capillary at a height of 30 μm above the cover slip. The spatial resolution of the measurement was affected by both the spot size and the step-size. The actual step-size represented a compromise between the scan time across 2000 pixels at 700 ms/pixel and the size of the multi-core optical fiber illuminating each pixel.
When excited by the pulsing blue LED, the ruthenium dye transfered energy by collisional quenching to the oxygen and the lifetime was reduced. With an appropriate calibration, the fluorescent lifetime measured by NeoFox can be related to the oxygen concentration using the Stern-Vollmer relationship. The quenching coefficient,  , depends on the viscosity η, which differed in hydrogel, tissue and medium. To account for the viscosity in the oxygen profile, two additional calibration scans of the fluorescent lifetimes were acquired: one for which the microfluidic was immersed in a <1% oxygen solution and equilibrated for 12 h; and another for which the microfluidic was immersed in ambient oxygen for 12 h. Temperature also affected the solubility of oxygen in samples, producing a change in the calibration slope. To avoid changes in temperature, the experiments were all conducted at 37.0 ± 0.1 °C.
, depends on the viscosity η, which differed in hydrogel, tissue and medium. To account for the viscosity in the oxygen profile, two additional calibration scans of the fluorescent lifetimes were acquired: one for which the microfluidic was immersed in a <1% oxygen solution and equilibrated for 12 h; and another for which the microfluidic was immersed in ambient oxygen for 12 h. Temperature also affected the solubility of oxygen in samples, producing a change in the calibration slope. To avoid changes in temperature, the experiments were all conducted at 37.0 ± 0.1 °C.
The sample was scanned with a 10 μm step-size and lifetimes were measured for 700 ms at each step. For a wide field, an area spanning 1240 × 620 μm2 was scanned yielding 1922 oxygen pixels, but typically data was acquired from smaller areas along and transverse to the capillary axis. At each point in the scan, a transmission image was captured as well. Finally, all transmission images were stitched together to form a transmission image and overlaid with the corresponding profile.
Finite Element Simulation of Oxygen transport
COMSOL Multiphysics 4.3 was used to perform finite element simulations of oxygen transport. The diffusion of oxygen was assumed to follow Fick’s laws and so, along with advection, the oxygen concentration throughout the capillary was modeled deterministically according to the equation:
 where [O2] is the local oxygen concentration and R denotes an oxygen sink (tissue), the velocity v was determined by another simulation using Navier-Stokes equation and incompressible flow with no-slip boundary conditions. The model consisted of a cylindrical tube with a lumen matching the average diameter along the capillary—ranging from 10 to 20 μm—situated 25 μm above the cover glass positioned in the center of a block 50 × 50 μm2, 200 μm long. The dimensions of the tube were taken from transmission images focused at the lumen acquired shortly before the oxygen measurements. The wall of the tube represented the hMVECs and hPPs. Another concentric tube 50 μm tall and 75 μm wide and as long as the capillary represented the hNFs. The remaining space between the walls of the microfluidic and cover-glass were filled with hydrogel except at the inlet and outlet where there was blood or medium. The diffusivity of dissolved oxygen, D, in the hydrogel polymer, media and blood was set to
where [O2] is the local oxygen concentration and R denotes an oxygen sink (tissue), the velocity v was determined by another simulation using Navier-Stokes equation and incompressible flow with no-slip boundary conditions. The model consisted of a cylindrical tube with a lumen matching the average diameter along the capillary—ranging from 10 to 20 μm—situated 25 μm above the cover glass positioned in the center of a block 50 × 50 μm2, 200 μm long. The dimensions of the tube were taken from transmission images focused at the lumen acquired shortly before the oxygen measurements. The wall of the tube represented the hMVECs and hPPs. Another concentric tube 50 μm tall and 75 μm wide and as long as the capillary represented the hNFs. The remaining space between the walls of the microfluidic and cover-glass were filled with hydrogel except at the inlet and outlet where there was blood or medium. The diffusivity of dissolved oxygen, D, in the hydrogel polymer, media and blood was set to  , based on a comparison of time-dependent measurements of the oxygen profile within those materials and water—no significant difference was observed73. In bulk tissue, the diffusivity was set to
, based on a comparison of time-dependent measurements of the oxygen profile within those materials and water—no significant difference was observed73. In bulk tissue, the diffusivity was set to  . For volumes containing hMVECs, hPPs and hNF, it was assumed that the cells acted as sinks consuming oxygen at a rate of 0.05 mol·m−3·s−1 in medium containing 0.5 mol·m−3. The boundary conditions were chosen to match the measurements. Accordingly, the inlet oxygen was set to 6 and 14 kPa partial oxygen and the oxygen 100 μm on the trans-side of the capillary was set to 1 and 7 kPa and the surfaces in contact with PDMS were set to 2 and 9 kPa, respectively for the capillaries of Fig. 5a,b. The glass bottom of the microfluidic devices was assumed to preclude any oxygen diffusion. For modeling, the 3D geometry of the capillaries were drawn and filled with pre-defined high-resolution grids.
. For volumes containing hMVECs, hPPs and hNF, it was assumed that the cells acted as sinks consuming oxygen at a rate of 0.05 mol·m−3·s−1 in medium containing 0.5 mol·m−3. The boundary conditions were chosen to match the measurements. Accordingly, the inlet oxygen was set to 6 and 14 kPa partial oxygen and the oxygen 100 μm on the trans-side of the capillary was set to 1 and 7 kPa and the surfaces in contact with PDMS were set to 2 and 9 kPa, respectively for the capillaries of Fig. 5a,b. The glass bottom of the microfluidic devices was assumed to preclude any oxygen diffusion. For modeling, the 3D geometry of the capillaries were drawn and filled with pre-defined high-resolution grids.
Additional Information
How to cite this article: Sarveswaran, K. et al. Synthetic Capillaries to Control Microscopic Blood Flow. Sci. Rep. 6, 21885; doi: 10.1038/srep21885 (2016).
References
Griffith, L. G. & Swartz, M. A. “Capturing complex 3D tissue physiology in vitro”, Nat. Rev Mol. Cell Biol. 7, 211 (2006).
Bhatia, S. N. & Ingber, D. E. “Microfluidic organs-on-chips”, Nat. Biotech. 32(4), 10.1038/nbt.2989 (2014).
Chen, C., Mrksich, M., Huang, S., Whitesides, G. & Ingber, D. “Geometric Control of Cell Life and Death”, Science 276, 1425–28 (1997).
Albrecht, D. R., Underhill, G. H., Wassermann, T. B., Sah, R. L. & Bhatia, S. N. “Probing the role of multicellular organization in three-dimensional microenvironments”, Nat. Meth. 3, 369–375 (2006).
Miller, J. S. et al. “Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues”, Nature Mat. 11, 768–774 (2012).
Lee, V. K. et al. “Generation of multi-scale vascular network system within 3D hydrogel using bio-printing technology”, Cell. Mol. Bioeng. 7, 460–472 (2014).
Hoch, E., Tovar, G. E. & Borchers, K. “Bioprinting of artificial blood vessels: current approaches towards a demanding goal”, Eur. J. Cardiothorac Surg. 46, 767–778 (2014).
Kirpatrick, C. J., Fuchs, S., Hermanns, M. I., Peters, K. & Unger, R. E. “Cell culture models of higher complexity in tissue engineering and regenerative medicine”, Biomat. 28, 5193–5198 (2007).
Seliktar, D. “Extracellular Stimulation in Tissue Engineering”, Ann. N.Y. Acad. Sci. 1047, 386–394 (2005).
Rivron, N. C., Liu, J., Rouwkema, J., De Boer, J. & van Blitterswijk, C. A. “Engineering Vascularised Tissues In Vitro”, Eur. Cells Mat. 15, 27–40 (2008).
Mirsaidov, U. et al. “Live Cell Lithography: Using Optical Tweezers to Create Synthetic Tissue”, Lab Chip, 8, 2174–2181 (2008).
Linnenberger, A. et al. “Three-dimensional live cell lithography”, Optics Express 21(8), 10269–277, (2013).
Fung, Y. C., Zweifach, B. W. & Intaglietta, M. “Elastic Environment of the Capillary Bed”, Circulation Research. 19, 441–461(1966).
Caro, C. G., Pedley, T. J., Schroter, R. C. & Seed W. A. In The Mechanics of the Circulation 2nd edn., Ch. 13, 346–425 (Cambridge University Press, 2012).
Liu, Y., Dang, C., Garcia, M., Gregersen, H. & Kassab, G. S. “Surrounding tissues affect the passive mechanics of the vessel wall: theory and experiment”, Am. J. Physiol. Heart Circ. Physiol. 293, H3290–H3300 (2007).
Hudetz, A. G. “Blood flow in the cerebral capillary network: a review emphasizing observations with intravital microscopy” Microcirculation 4(2), 233–52 (1997).
DeCicco-Skinner, K. L. et al. “Endothelial Cell Tube Formation Assay for the In Vitro Study of Angiogenesis”, J. Vis. Exp. 91, e51312 (2014).
Kurz, V., Tanaka, T. & Timp, G. “Single Cell Transfection with Single Molecule Resolution Using a Synthetic Nanopore”, Nano Lett. 14(2), 604–611 (2014).
Lutolf, M. P. & Hubbell, J. A. “Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering”, Nat. Biotech. 23(1), 47–55 (2005).
Ramo΄n-Azco΄n, J. et al. “Gelatin methacrylate as a promising hydrogel for 3D microscale organization and proliferation of dielectrophoretically patterned cells”, Lab Chip 12, 2959–2969 (2012).
Turturro, M. V. et al. “MMP-Sensitive PEG Diacrylate Hydrogels with Spatial Variations in Matrix Properties Stimulate Directional Vascular Sprout Formation”, PLoS One. 8(3), e58897 (2013).
Zhu, Y., Dong, Z., Wejinya, U. C., Jin, S. & Ye, K. “Determination of mechanical properties of soft tissue scaffolds by atomic force microscopy nanoindentation”, J. Biomechanics 44, 2356–61 (2011).
Drira, Z. & Yadavalli, V. K. “Nanomechanical measurements of polyethylene glycol hydrogeles using atomic force microscopy”, J. Mech. Behavior Biomedical Mat. 18, 20–28 (2013).
Zeng, D. et al. “Young’s modulus of elasticity of Schlemm’s canal endothelial cells”, Biomech Model Mechanobiol. 9, 19–33 (2010).
Solon, J., Levental, I., Sengupta, K., Georges, P. C. & Janmey, P. A. “Fibroblast Adaptation and Stiffness Matching to Soft Elastic Substrates”, Biophysical J. 93, 4453–4461 (2007).
Rojkind, M. & Perez-Tamayo, R. “Liver Fibrosis”, Int. Rev. Connect. Tissue Res. 10, 333–393 (1983).
Kara, M., Ragbetli, C., Koyun, N., Colcimen, N. & Ragbetli, M. C. “The stereological ratio of stromal/parenchymal components in postnatal prostate tissue of rats prenatally treated with diclofenac sodium”, J. Exp. Clin. Med. 30, 171–174 (2013).
Briscoe, B. J., Sebastian, K. S. & Adams, M. J. “The effect of indenter geometry on the elastic response to indentation”, J. Phys. D: Appl. Phys. 27, 1156–1162 (1994).
Raeber, G. P., Lutolf, M. P. & Hubbell, J. A. “Molecularly Engineered PEG Hydrogels: A Novel Model System for Proteolytically Mediated Cell Migration”, Biophys. J. 89, 1374–1388 (2005).
Zhao, X., Hong, W. & Suo, Z. “Inhomogeneous and anisotropic equilibrium of a swollen hydrogel containing a hard core”, Appl. Phys. Lett. 92, 051904 (2008).
Olcuma, S. et al. “Weighing nanoparticles in solution at the atto-gram scale”, Proc. Nat. Acad. Sci. USA 111(4), 1310–1315 (2013).
Armulik, A., Abramsson, A. & Betsholtz, C. “Endothelial/Pericyte Interactions”, Circ. Res 97, 512–523 (2005).
Berthod, F., Germain, L., Tremblay, N. & Auger, F. A. “Extracellular matrix deposition by fibroblasts is necessary to promote capillary-like tube formation in vitro”, J Cell Physiol. 207, 491–498 (2006).
Ghajar, C. M. et al. “The effect of matrix density on the regulation of 3-D capillary morphogenesis”, Biophys. J. 94(5), 1930–1941 (2008).
Landau, J. & Davis, E. “The small blood-vessels of the conunctiva and nailbed in diabetes mellitus”, Lancet 269, 1327–1330 (1957).
Starling, E. H. Principles of Human Physiology, Ch. 13, 979–1129 (Lea and Febiger, 1912).
Cleaver, O. & Melton, D. G. “Endothelial signaling during development”, Nat. Med. 9, 661–8 (2003).
Rhodin, J. A. “Ultrastructure of mammalian venuous capillaries, venules and small collecting veins”, J Ultrastruct Res. 25, 452–500 (1968).
Bedggood, P. & Metha, A. “Direct visualization and characterization of erythrocyte flow in human retinal capillaries,” Biomed. Optics Express 3, 3264–3277 (2012).
Suter, E. R. & Majno, G. “Passage of lipid across vascular endothelium in newborn rats: an electron microscopic study”, J Cell Biol. 27, 163–177 (1965).
Casley-Smith, J. R., Green, H. S., Harris, J. L. & Wadey, P. J. “The quantitative morphology of skeletal muscle capillaries in relation to permeability”, Microvascular Res. 10, 43–64 (1975).
Wisse, E. “An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids”, J. Ultrastructure Res. 31, 125–150 (1970).
Yeon, J. H., Ryu, H. R., Chung, M., Hu, Q. P. & Jeon, N. L. “In vitro formation and characterization of a perfusable three-dimensional tubular capillary network in microfluidic devices”, Lab Chip 12(16), 2815–22 (2012).
Zheng, Y. et al. “In vitro microvessels for the study of angiogenesis and thrombosis”, Proc. Nat. Acad. Sci USA 109(24), 9342–7 (2012).
Hsu, Y.-H., Moya, M. L., Hughes, C. C., George, S. C. & Lee, A. P. “A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays”, Lab Chip 13(15), 2990–8 (2013).
Moya, M. L., Hsu, Y.-H., Lee, A. P., Hughes, C. C. & George, S. C. “In vitro perfused human capillary networks”, Tissue Engineering: Part C. 19(9), 730–7 (2013).
Moya, M. L., Alonzo, L. F. & George, S. C. “Microfluidic Device to Culture 3D In Vitro Human Capillary Networks”, Methods in Molecular Biology 1202, 21–7 (2014).
Verbridge, S. S. et al. “Physicochemical regulation of endothelial sprouting in a 3D microfluidic angiogenesis model”, J. Biomed. Mater. Res. 101A, 2948–2956 (2013).
Jain, R. K. “Molecular regulation of vessels maturation”, Nat. Med. 9, 68 (2003).
Davis, G. E., Stratman, A. N., Sacharidou, A. & Koh, W. “Molecular basis for endothelial lumen formation and tubulogenesis during vasculogenesis and angiogenic sprouting. Int Rev Cell Mol Biol. 288, 101–165 (2011).
Helm, C. E., Zisch, A. & Swartz, M. A. “Engineered Blood and Lymphatic Capillaries in 3-D VEGF-Fibrin-Collagen Matrices with Interstitial Flow”, Biotech Bioeng. 96(1), 167–176 (2007).
Kaully, T., Kaufman-Francis, K., Lesman, A. & Levenberg, S. “Vascularization: The conduit to Viable Engineered Tissue”, In Advances in Tissue Eng: Angiogenesis (eds. Johnson P. C. & Mikos A. G. ) 17–26 (Mary Ann Liebert, Inc., 2010).
Tian, L. & George, S. C. “Biomaterials to Prevascularize Engineered Tissues”, J. Cardiovasc. Trans. Res. 4, 685–698 (2011).
Tkachenko, E. et al. “The nucleus of endothelial cells as a sensor of blood flow”, Biology Open 2, 1007–1012 (2013).
Lamberti, G. et al. “Adhesion patterns in the microvasculature are dependent on bifurcation angle”, Microvascular Res. 99, 19–25 (2015).
Koutsiaris, A. G. et al. “Volume flow and wall shear stress quantification in the human conjunctival capillaries and post-capillary venules in vivo”, Biorheology. 44, 375–386 (2007).
Greve, J. M. et al. “Allometric scaling of wall shear stress from mice to humans: quantification using cine phase-contrast MRI and computational fluid dynamics. Am. J. Physiol. Heart Circ. Physiol. 291, H1700–H1708 (2006).
Song, J. W. & Munn, L. L. “Fluid forces control endothelial sprouting”, Proc. Nat. Acad. Sci. 108(37), 15342–15347 (2011).
Radisic, M. et al. “Oxygen Gradients Correlate With Cell Density and Cell Viability in Engineered Cardiac Tissue”, Biotech. Bioeng. 93(2), 332–343 (2006).
Buchwald, P. “FEM-based oxygen consumption and cell viability models for avascular pancreatic islets”. Theoretical Biology Med. Model. 6, 5 (2009).
Kellner, K. et al. “Determination of oxygen gradients in engineered tissue using a fluorescent sensor”, Biotech. Bioeng. 80(1), 73–83 (2002).
Malda, J. et al. “Oxygen gradients in tissue-engineered Pegt/Pbt cartilaginous constructs: Measurement and modeling”, Biotech. Bioeng. 86(1), 9–18 (2004).
Federspiel, W. J. & Popel, A. S. “A Theoretical Analysis of the Effect of the Particulate Nature of Blood on Oxygen Release in Capillaries”, Microvascular Res. 32, 164–189 (1986).
Strell, C. & Entschladen, F. “Extravasation of leukocytes in comparison to tumor cells”, Cell Commun. Signal. 6(10), 10.1186/1478-811X-6-10 (2008).
Kienast, Y. et al. “Real-time imaging reveals the single steps of brain metastasis formation”, Nat. Med. 16(1), 116–123 (2010).
Lee, H., Park, W., Ryu, H. & Jeon N. L. “A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation”, Biomicrofluidics 9(5), 054102 (2014).
Zervantonakis, S. K. et al. “Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function”, Proc. Nat. Acad. Sci. USA 109(34), 13515–20 (2012).
Parikh, M. R., Minser, K. E., Rank, L. M., Glackin C. A. & Kirshner, J. “A reconstructed metastasis model to recapitulate the metastatic spread in α”, Biotech. J. 9, 1129–39 (2014).
Ioachim, E. et al. “Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression.” Eur. J. Cancer. 38, 2362–2370 (2002).
Akselrod, G. et al. “Laser-Guided Assembly of Heterotypic Three-Dimensional Living Cell Microarrays”, Biophys. J. 91, 3465–3473 (2006).
Nichol, J. W. et al. “Cell-laden microengineered gelatin methacrylate hydrogels”. Biomaterials. 31, 5536–44 (2010).
Buxton, A. N. et al. “Design and Characterization of Poly(Ethylene Glycol) Photopolymerizable Semi Interpenetrating Networks for Chondrogenesis of Human Mesenchymal Stem Cells”, Tissue Eng. 13(10), 2549–2560 (2007).
Han, P. & Bartels, D. M. “Temperature Dependence of Oxygen Diffusion in H2O and D2O”, J. Phys. Chem. 100, 5597 (1996).
Acknowledgements
This work was supported by the Keough-Hesburgh professorship, KS received support from Advanced Diagnostic and Therapeutics at Notre Dame. SP received partial support from NDNano at Notre Dame. VK received partial support from the Walther Cancer ENCCII Training Project (#261799-28110-62010-20000-11104) obtained through the Harper Cancer Institute.
Author information
Authors and Affiliations
Contributions
G.T., V.K., T.T., K.S. and Z.D. wrote the manuscript. G.T., T.T., K.S. and V.K. conceived and planned the experiments. Koshala, Z.D. and V.K. acquired the data. K.S., V.K., G.T. and S.P. analyzed the data. G.T. and K.S. prepared Figures 1,2 and 3; G.T. and Z.D. prepared Figures S1, S2 and S3; G.T., K.S., S.P. and V.K. prepared Figures 4 and 5; G.T., K.S. and T.T. prepared Figure 6. All the authors reviewed the manuscript.

Schematic of a time-shared optical trapping apparatus and microfluidic conveyer used to form a synthetic capillary.
(a) Cells were manipulated using optical traps formed with infrared light (red path) from a Ti:sapphire laser through a high NA commercial optical microscope. The same optics were used for imaging (yellow path) and near-UV exposure to photopolymerize the hydrogel scaffold (blue path). Two or more cell types were conveyed to the assembly area using the microfluidic network. A hydrogel scaffold was formed in the cross-channel. (a, upper insets) micrographs of a 3-layer structure of hMVECs organized using tweezers to resemble a capillary and then encapsulated in a hydrogel scaffold. (a, lower inset) perspective rendering of the entire microfluidic device. (b) Bar-graphs are shown that relate the viability scores for various cell types 10 h after manipulation with optical tweezers and encapsulation in hydrogel. Three hundred eighteen hMVECs (gray), 246 hNFs(orange) and 438 hPPs (blue) were assayed after exposure. They were exposed to time-shared optical traps at wavelengths of 800, 830, 850 and 870 nm with 0, 50, 100 and 200 mW per cell. The cells were held in the trap for 30 sec prior to gelling in a PEGDA hydrogel and then viability was assessed with LIVE/DEAD stains.

Layer-by-layer assembly of a synthetic capillary.
(a–e, left) Top-down optical micrographs of a 3-layer structure of hMVECs (endothelials) and hPPs (pericytes) partially encapsulated in PEGDA hydrogel is shown, illustrating the basement layer, the lumen and the attic layer respectively, which was organized using optical tweezers to resemble a capillary about one-quarter millimeter long. The optical micrographs show living voxels each containing a single cell encapsulated in a hydrogel scaffold. Selected hPPs are indicated by the arrows in (d), whereas the final capillary encased in voxels constituted from hNFs (fibroblasts) in a Gel-MA scaffold are represented in (e) with selected hNFs indicated by the arrows. (a–e, right) Schematic representations are shown of the construction with hMVECs denoted by dark gray circles, pericytes by light gray and fibroblasts by medium gray. (f) A false-color perspective shadow projection is shown, which was reconstructed from volumetric data acquired from a series of confocal images from IF stained cells to differentiate hMVECs from hPPs and hNFs. As the fluorescent intensity is mapped using shadow projection, the fluorescent surface is visually impenetrable. (g,h) Two 20 μm thick slabs are shown, which were reconstructed from volumetric (confocal) data showing cross-sectional (through the lumen) views in the same capillary. To visualize the intraluminal surfaces, the hMVECs were positively stained for CD31 (green), whereas the hPPs and hNFs were positively stained for NG2 (red). (g, upper right inset) A thin 0.5 μm cross-section through the same capillary, stained with an FITC-conjugated anti-CD31 antibody, is shown. Cells exhibit localization of CD31 predominantly on the cell surface, whereas localization out of the plane (arrows) was not obvious.

Remodeling in a capillary and a branched arteriole created with LCL.
(a) Time-lapse optical micrographs are shown, which were taken every 30 min show remodeling in a synthetic capillary. In particular, the images of the lumen acquired at 30 min and 6 h are shown. The white arrows, which are in fixed positions in the two images, highlight areas where cells are remodeling. (b) A top-down optical micrograph of a multi-layer structure of hMVECs and hVSMCs, partially encapsulated in PEGDA/PEG-RGDS hydrogel, is shown, which was organized using optical tweezers to resemble a branched arteriole about 0.33 mm long. Selected hSMCs and hMVECs are indicated by the arrows. (c) A false-color perspective shadow projection is shown, which was reconstructed from volumetric data acquired from a series of confocal images from immunofluorescently stained CD31-postive cells or the arteriole in (b). (Top and bottom insets) Cross-sectional views (through the lumen) of the arteriole in (b) before (top) and after (bottom) the bifurcation are shown, which are positioned as indicated by the yellow and white dotted rectangles on the left, respectively.

Erythrocytes (red blood cells) perfusing through a synthetic capillary.
(a) False color representation of erythrocytes flowing through a capillary are shown, which were obtained from a series of confocal slices through the lumen of the vasculature acquired every 16 ms. These snapshots shown here were taken at 48 ms intervals. The erythrocyte trajectory is indicated by the white solid line. In this case, the velocity of the cell, estimated from the distance an erythrocyte traveled between frames, was 586 ± 6 μm/s. (b) Like (a), but a superposition of the positions of ten erythrocytes as they flow through the capillary. (c) A confocal slice through the vasculature like that shown was used to estimate the cross-sectional area of the lumen. Fluorescent microspheres 40 nm in diameter were used to define the lumen as a function of position along the axis of the capillary. (d) Histogram of the axial velocity versus the position in the lumen is shown, which was obtained from measurements of >1000 erythrocytes. The dotted red line is the best-fit parabola; the deviation is an indication of a non-Newtonian fluid. (e) A summary of the measurements of the axial velocity as a function of the cross-sectional area of the lumen is shown, which was obtained for two capillaries: one with a diameter <10 μm at differential pressures of 0.1, 0.5 and 1.0 kPa (color); and another with a diameter <20 μm at differential pressures of 0.1, 1.5 and 2.5 kPa (grey). The lines, which represent power-law fits to the data, act as guides to the eye. (f) Like (e), but a corresponding summary of the wall shear stress (WSS) on the same capillaries as a function of the cross-sectional area inferred from the axial velocity with pressure as a parameter.

Oxygen flow through and gradients across a synthetic capillary.
(a,b, left) Tiled transmission images of two synthetic capillaries embedded in a microfluidic device are shown. The lumen was assembled from HMVECs and hPPs, whereas the abluminal cells were hNFs. Rectangular PDMS supports (corners) were used for mechanical support. The capillary in (a) has a lumen with a 100 μm2 cross-sectional area and experienced a flow of 0.25 μl/min, whereas the capillary in (b) had a 400 μm2 lumen and was subject to a 0.05 μl/min flow. (a,b, center) Reconstructed 2D heat map of the oxygen profile is shown, which was acquired from the structures on the left. The measurements of the oxygen concentration were accomplished in 10 μm steps. (a,b,right) FESs of the oxygen flow through an idealized capillary under flow conditions similar to that used for the structures on the left is shown, which takes into account, diffusion, migration and consumption of oxygen molecules by the cells. The partial pressure of dissolved O2 across the endothelial cell wall was about 15 kPa regardless of the differential pressure across the capillary. (c,d) The oxygen gradients measured along the capillary axes and in the center transverse to them are shown. The (top,bottom) solid lines correspond to measurements extracted from the capillaries in (a,b) respectively, the center solid line was associated with a capillary shown in the supplement (Fig. S6). The dotted lines represent the results of FES.

A metastatic human breast cancer cell trapped in a synthetic capillary.
Time-lapse optical micrographs taken every 10 min are shown, which illustrate the progress of a single MDA-MB-231 cancer cell (highlighted in red) over 4 h. The hMVECs that contact the cancer cells are indicated with asterisks.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sarveswaran, K., Kurz, V., Dong, Z. et al. Synthetic Capillaries to Control Microscopic Blood Flow. Sci Rep 6, 21885 (2016). https://doi.org/10.1038/srep21885
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21885
This article is cited by
-
Development of Tumor-Vasculature Interaction on Chip Mimicking Vessel Co-Option of Glioblastoma
BioChip Journal (2023)
-
Engineered models to parse apart the metastatic cascade
npj Precision Oncology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
