
Abstract
Plant evolutionary history influences the taxonomic composition of the root-associated bacterial community, but whether it can also modulate its functioning is unknown. Here, we tested the hypothesis that crop diversification is a significant factor determining the ecology of the functional group of nitrogen-fixing bacteria the rhizosphere of Poaceae. A greenhouse experiment was carried out using a range of Poaceae, i.e. four Zea mays varieties (from two genetic groups) and teosinte (representing maize’s ancestor), sorghum (from the same Panicoideae subfamily) and wheat (from neighboring Pooideae subfamily), as well as the dicot tomato as external reference. Diazotroph rhizosphere community was characterized at 21 days in terms of size (quantitative PCR of nifH genes), composition (T-RFLP and partial sequencing of nifH alleles) and functioning (quantitative RT-PCR, T-RFLP and partial sequencing of nifH transcripts). Plant species and varieties had a significant effect on diazotroph community size and the number of nifH transcripts per root system. Contrarily to expectations, however, there was no relation between Poaceae evolutionary history and the size, diversity or expression of the rhizosphere diazotroph community. These results suggest a constant selection of this functional group through evolution for optimization of nitrogen fixation in the rhizosphere.
Similar content being viewed by others


Introduction
Nitrogen is essential for plant growth and health. However, it is not always bioavailable in soil, as most nitrogen is present as N2 in soil porosity or as part of humic compounds. Amongst prokaryotes, diazotrophs can facilitate plant nitrogen nutrition through biological nitrogen fixation, which corresponds to the reduction of atmospheric nitrogen (N2) to ammonia (NH3). This occurs in agricultural as well as in natural environments and amounts to 120 million tons of N are fixed annually on earth1. Biological nitrogen fixation is only performed by certain bacterial and archaeal taxa and all rely on the nifH gene encoding the iron protein subunit of the nitrogenase, a gene widely used as marker to examine nitrogen fixation in various environments2,3,4. This gene is highly conserved among diazotrophs and its phylogeny is largely correlated to 16S rRNA phylogeny5, making it a marker suitable to assess complex diazotrophic communities.
In terrestrial systems, diazotrophic community size and/or composition can vary according to soil type3,6,7,8, soil management9, season7, fertilization rate2,10, plant presence/absence2,11,12, plant growth stage3,12, plant species13,14,15,16 or plant varieties2,17. Some of these factors may also influence the composition of the bacterial community actually expressing nifH and/or the amount of nifH transcripts, even if the latter does not necessarily vary to a large extent18,19,20,21.
Differences in root-associated nitrogen-fixing bacterial communities between plant genotypes may involve differences in (i) root system development22 and thus bacterial root-colonization sites, (ii) nutrient uptake by roots23 and therefore rhizosphere depletion of minerals including nitrate and ammonium and (iii) root exudation patterns24 and thus the flux of carbon sources and energy available to support BNF. However, plant genetic properties accounting for these plant phenotypic differences are not well understood and those determining differences in root-associated functional microbial groups are even less understood. Evolutionary processes, whether corresponding to natural selection and/or human selection in agrosystems, have resulted in a wide range of plant genotypes on earth25 and have had a strong influence on root traits23,26,27,28. Recently, it was shown that the evolutionary history of Poaceae grown in a same soil was a significant factor determining the taxonomic composition of the total rhizobacterial community29. This was also the case at the level of several bacterial genera containing nitrogen-fixing species or strains, raising the possibility that root-associated microbial functional groups, such as nitrogen-fixing bacteria, may also be influenced by the evolutionary history of Poaceae. On this basis, our hypothesis was that plant evolutionary history can be a significant factor influencing the interaction of roots with microbial functional groups.
The objective of this study was to assess whether a relation exists between Poaceae evolution and root-associated diazotroph community of the resulting plant genotypes. To this end, we compared Poaceae both at infraspecific (four lines from two contrasted genetic groups of maize and one teosinte representing maize’s pre-domestication ancestor) and interspecific levels (one sorghum from maize’s Panicoideae subfamily and one wheat from neighboring Pooideae subfamily) in a same soil under greenhouse conditions. A dicot (tomato) was also included as non-Poaceae external reference. The diazotroph community was characterized at 21 days in terms of size (nifH quantitative PCR), composition (nifH T-RFLP and partial sequencing of nifH gene) and functioning (quantitative RT-PCR, T-RFLP and partial sequencing of nifH transcripts). For selected treatments, the analyses were also carried out with better-established plants (i.e. at 42 days after sowing) as well as with 21-day-old plants grown in soil from a neighboring field (from the same soil type but under permanent meadow instead of maize monocropping), because in both cases rhizobacterial community structure and taxonomic composition differed29.
Results
Size of diazotroph communities
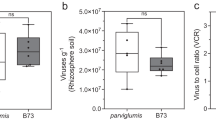
The total nitrogen-fixing bacterial community amounted to 1.0–4.6 × 107 nifH gene copies per g of soil (Fig. 1A) and for plant treatments it meant 1.1–11.0 × 108 nifH copies per g of root (Fig. 1C) or 4.8–260 × 108 nifH copies per root system (Fig. 1E). At 21 days in cropped soil, diazotroph community size per g of soil was higher in the presence of plant for all maize lines and for wheat (Fig. 1A). This was also the case for 2 of 3 plant genotypes at 42 days in the same soil, whereas differences were not significant at 21 days in meadow soil.

Quantification of nifH genes and transcripts in bulk soil and rhizosphere of Poaceae genotypes by real-time PCR and RT-PCR, respectively.
Gene copy numbers are shown in (A,C,E) and transcript numbers in (B,D,F). Statistical analyses were performed independently at 21 days in cropped soil, at 21 days in meadow soil and at 42 days in cropped soil, using ANOVA and Fisher LSD tests (P < 0.05; results shown with letters a to (D). For FV4, W85, tomato and bulk soil, two-way ANOVA and Fisher LSD tests (P < 0.05) were also performed to compare treatments according to past soil management or sampling time and differences with the same genotype at 21 days in cropped soil are indicated by symbols *and #respectively.
When expressed per g of root, the diazotroph community size at 21 days in cropped soil did not differ for maize line FV252, teosinte, sorghum and tomato, whereas wheat and the two Northern Flint maize lines gave higher levels (Fig. 1C). At 21 days, differences (but not the same as in cropped soil) between plant genotypes were also found in meadow soil, whereas these differences were not significant at 42 days in cropped soil.
Since root system development varied according to plant genotype (as well as sampling time and past soil management for maize line FV4) (Figure S1A), diazotroph community size was also considered per root system. When doing so (Fig. 1E), diazotroph community at 21 days in cropped soil did not differ in size for teosinte, wheat, sorghum, whereas its size was higher for maize lines W85 and Mo17 and lower for tomato. nifH copy number was also lower for tomato than for maize at 21 days in meadow soil, whereas differences were not significant at 42 days in cropped soil.
Overall, differences in diazotroph community size were found according to plant genotype, sampling time or past soil management. Even though differences occurred between plant genotypes, more closely related plant genotypes did not necessarily display a diazotroph community closer in size.
nifH transcript levels in diazotroph communities
The number of nifH transcripts from the nitrogen-fixing bacterial community reached 1.3–4.8 × 104 nifH cDNA copies per g of soil (Fig. 1B) and for plant treatments it corresponded to 1.4–8.9 × 105 nifH cDNA copies per g of root (Fig. 1D) or 5.4–910 × 103 nifH cDNA copies per root system (Fig. 1F). At 21 days in cropped soil, the number of nifH transcripts per g of soil was similar in all treatments (Fig. 1B). This was also the case in the same soil when considering the other sampling (42 days), or at 21 days in meadow soil.
When focusing on plant treatments and expressing data per g of root, regardless of the amount of rhizosphere soil (Figure S1B), there was again no difference between genotypes in the number of nifH transcripts in cropped soil at 21 or at 42 days (Fig. 1D). In meadow soil at 21 days, however, the number of nifH transcripts per g root was higher for the two maize lines than for tomato.
When integrating root system size, the number of nifH transcripts (i.e. expressed per root system) at 21 days in cropped soil was higher for maize lines W85 and Mo17 than for maize lines FV4 and FV252, wheat and tomato (Fig. 1F). At 21 days in meadow soil, the number of nifH transcripts per root system was again higher for maize line W85 than for tomato, but this time also maize line FV4 gave higher values than tomato. There was no difference in cropped soil at 42 days, when tomato root system was larger.
Overall, differences in nifH transcript number were mainly found when comparing plant treatments using data expressed per root system (i.e. at the scale of entire rhizospheres), but plant genotypes from the same maize group or species did not give values more similar than did genotypes from different species.
Diversity of diazotroph communities
T-RFLP of bulk soil and rhizosphere treatments revealed a total of 43 distinct T-RFs (from 30 bp to full 350-bp polF/polR amplicon size), of which 21T-RFs were found only in DNA samples, 9 in cDNA samples and 13 in both (Fig. 2). With DNA samples, 3T-RFs were evidenced in at least 10 of 17 treatments, 8 in 6–9 treatments, 18 in 2–5 treatments and 8 in only 1 treatment. With cDNA samples, 2T-RFs were evidenced in 6–9 treatments, 8 in 2–5 treatments and 12 in only 1 treatment. The genetic distance between samples was higher (meaning a lower profile variability) for DNA samples (0.53 ± 0.01) than for cDNA samples (0.46 ± 0.01).

Comparison of T-RFLP profiles from bulk soil and rhizosphere of Poaceae genotypes based on nifH DNA and cDNA samples in cropped soil at 21 days (black circles) and 42 days (white circles) after sowing and in meadow soil at 21 days (black triangles).
The analysis was based on presence/absence of T-RFs in each treatment, using Euclidean distance matrix and complete linkage clustering. The two T-RFs evidenced by in-silico T-RFLP of nifH qPCR and qRT-PCR products from W85 maize correspond to peaks 1 and 2.
Clustering of all treatment combinations based on T-RFLP data gave 3 clusters (C1–C3) as well as, separately, the bulk soil sample from cropped soil at 42 days for DNA samples and 3 other clusters (C4–C6) for cDNA samples (Fig. 2). When considering past soil management, cropped soil samples clustered with meadow soil samples for bulk soil DNA (in C3), maize FV4 DNA (in C3) and maize FV4 cDNA (in C5), which was not the case for bulk soil cDNA (C4 vs C5), maize W85 DNA (C1 vs C3) and cDNA (C4 vs C6), or tomato DNA (C2 vs C3) and cDNA (C4 vs C5). Cropped soil samples at 21 days clustered with the ones at 42 days for maize FV4 DNA (in C3) and maize W85 DNA (in C1), but not for the other DNA samples or any of the cDNA samples, even in the case of bulk soil. Against this background, the two maize lines FV252 and Mo17 of the genetic group Corn Belt Dent clustered together both for DNA (in C2) and cDNA (in C5). However, it was not the case with the two Northern Flint maize lines (for DNA or cDNA) and overall the four maize lines were split in all three DNA clusters and all three cDNA clusters. No particular relation was found either when adding successively the fifth Zea mays genotype (teosinte), sorghum and wheat, regardless of whether cDNA or DNA was considered.
Sequencing the qPCR and qRT-PCR products from the W85 sample studied gave 70 and 180 partial nifH sequences, respectively, which gave 14 clades and 3 single sequences (Figure S2). Six of the 14 clades contained nifH sequences from known taxa, i.e. Burkholderia/Curvibacter (clade 2), Ideonella/Azohydromonas/Ensifer (clade 8), Rubrivivax (clade 9), Azorhizobium/Bradyrhizobium (clade 10), Azospirillum (clade 12) and Rhizobium/Sinorhizobium (clade 13), whereas the eight others did not. Except for clade 10, each clade included both DNA and RNA sequences. In silico T-RFLP analyses of the 250 nifH DNA and cDNA sequences gave only 2 distinct T-RFs, which correspond to T-RFs actually obtained by T-RFLP of nifH qPCR and qRT-PCR products.
Relationship between Poaceae phylogeny and diazotroph community
For a more formal appraisal of the relationship between diazotroph community and Poaceae phylogeny, correlation analysis was carried out between pairwise Poaceae phylogenetic distances and Euclidean distances derived from their root-associated diazotroph communities at 21 days in cropped soil (Fig. 3). A significant correlation was found when considering nifH gene copy number per g of root (r = 0.60, P = 0.004, n = 18; Fig. 3E) but not per g of rhizosphere soil or per root system (Fig. 3C,G), as well as nifH transcript number per g of rhizosphere soil (r = 0.59, P = 0.004, n = 18; Fig. 3D) but not per g of root or per root system (Fig. 3F,H). No significant correlation was obtained when considering diazotroph community diversity, regardless of whether data were derived from DNA or cDNA T-RFLP analysis (Fig. 3A,B).

Pairwise comparisons (n = 18) of plant phylogenetic distance between Poaceae (X axis) with the corresponding distance between their root-associated diazotroph communities (Y axis).
nifH DNA data are shown to the left and nifH cDNA data to the right and they correspond to results of T-RFLP (A,B), quantitative PCR or RT-PCR expressed per g of soil (C,D), per g of root (E,F) or per root system (G,H). Distances were calculated two by two, using Kimura 2 parameter model for plant phylogeny and Euclidean distance for diazotrophic community. The Pearson correlation coefficient and its probability level (when P < 0.05) are indicated.
Relationship between plant development and diazotroph community
A significant correlation was found when assessing plant phylogenetic distance against differences in plant development (in terms of shoot or root biomass) but not rhizosphere size (Table 1). Differences in the number of nifH gene copies correlated with differences in (i) shoot and root biomass (when expressed in copies per g of root), (ii) root biomass and rhizosphere mass (when expressed in copies per g of rhizosphere soil) and (iii) rhizosphere mass (when expressed in copies per root system). In addition, differences in the number of nifH transcripts correlated with differences in shoot biomass, root biomass and rhizosphere mass when expressed in copies per root system. Conversely, there was no correlation between (i) differences in shoot biomass, root biomass and rhizosphere mass and (ii) distances between T-RFLP profiles for nifH genes or transcripts.
Discussion
In this study, the impact of plant evolutionary history on root-associated diazotroph community size, nifH transcript number and genetic diversity was studied using molecular approaches based on established nifH primers9,20. The choice of primers is critical to capture the diversity of a functional group. Different nifH primer sets were designed in the last two decades and their potential to recover the majority of known nifH alleles was evaluated several times, with contradictory results30. When assessed in silico, primers such as the Zf/Zr pair31 presented a higher theoretical recovery of nifH diversity than the polF/polR primers used here32, but polF/polR display higher performance9,20 in vitro than in silico and can be used directly for qPCR (unlike others that are less specific9,20,30 and could require nested PCR31).
In the case of nitrogen-fixing rhizobia, the interaction with the plant (Fabales) leads to nodule structural differentiation34 and provides mutual benefits to the partners35. Most other plant families do not engage in this type of symbiosis, but in soil a large range of free-living microorganisms interact with plant roots, providing here also mutual benefits36,37,38. Although no partner differentiation takes place, it makes sense that during evolution, modifications of root phenotypic properties facilitating bacterial nitrogen fixation and thereby the supply of available nitrogen to the plant could have been selected.
On the one hand, many plant-beneficial free-living bacteria can benefit plants, but with contrasted efficacy according to plant species or varieties39,40. On the other hand, Poaceae evolutionary history is a factor influencing root selection of soil bacteria and thus total rhizobacterial community composition29. This concerns in particular many genera and species of nitrogen-fixing bacteria from the orders Rhodospirillales, Burkholderiales and Enterobacteriales. However, even though a significant relation was found in Bouffaud et al.29 between total rhizobacterial community composition and the phylogeny of the same Poaceae genotypes used in this study, no relation was found here when considering diazotroph community composition (Fig. 3). One such correlation was found with nifH gene copy number and another with nifH transcript number, but their relevance is probably limited because (i) the former was significant when nifH gene copy number was expressed per g of root but not per g of rhizosphere soil and the latter when nifH transcript number was expressed per g of rhizosphere soil but not per g of root, (ii) none of the two was significant when these community parameters were expressed per root system, which represents best the nitrogen fixation potential for plant individuals and (iii) the two correlation coefficients were of modest level (≤0.60). On this basis, it can be concluded that Poaceae evolution did not influence significantly nifH community size, the quantity of nifH transcripts or genetic diversity of root-associated nitrogen-fixing community.
Two features might account for the lack of relation found between Poaceae evolution and root-associated nitrogen-fixing community. First, results showed nifH bacteria were well present and expressed nifH in bulk soil, which means that presence of the plant was not required for their establishment and functioning (Fig. 1), thereby limiting the potential impact of plant features on the diazotroph community. Second, the relation between Poaceae evolution and rhizobacterial community composition found by Bouffaud et al.29 with the same seven plant genotypes was relevant for only one third (91 probes of 298) of bacterial taxa, which could mean that the housekeeping plant markers that were used did not reflect sufficiently the whole scope of plant traits relevant for plant-bacteria interactions. Another third (51 probes) of the 150 most discriminant 16S rRNA probes when comparing the taxonomic composition of the total rhizobacterial community29 targeted diazotrophic bacteria. The signal of 31 of these 51 diazotroph probes correlated in fact with maize/Poaceae genetic distances (e.g. for Azospirillum, Gluconobacter, Paenibacillus, etc.). However, it was not the case for the 20 other probes (e.g. for Bradyrhizobium, Devosia, etc.), which did not enable to yield a significant relation at the scale of the whole diazotrophic functional group.
Although Poaceae evolution was not a significant factor, an impact of plant genotype was yet found on diazotroph community size, expression or diversity. The impact on qPCR and qRT-PCR levels was rather moderate in magnitude, in accordance with previous studies showing differences usually not exceeding one log for DNA or cDNA copy number between samples from different areas or management practices15,20,21,41,42,43. This could suggest that a basal level of diazotrophic community size and expression always occurs (as discussed above for bulk soil) and is not extensively affected by environmental factors. In comparison, shifts in diazotrophic community populations occur readily20, including when comparing plant genotypes as found here (Fig. 2) and in previous work2,12,14,17. However, there was no apparent pattern of diazotrophic community structuration when considering genotypic groups defined within the maize species or across different plant species. Against this background, significant relations were found between plant development parameters and nifH gene or transcript numbers, suggesting that plant development could be a more important factor than plant evolutionary history and that occasional relations evidenced between Poaceae evolutionary history and nifH parameters probably involved Poaceae evolutionary history effects on plant development parameters.
In conclusion, this study showed that the effect of Poaceae evolutionary history on the root-associated diazotroph community was not significant, contrarily to expectations derived from the observation29 that plant evolutionary history did influence the taxonomic composition of the entire rhizobacterial community of Poaceae.
Materials and methods
Plant genotypes and greenhouse experiment
Plant genotypes included four cultivated maize (Zea mays L.), i.e. the inbred lines FV4, W85 (group Northern Flint), FV252 and Mo17 (group Corn Belt Dent) (provided by INRA, St Martin d’Hynx, France), one teosinte (Zea mays ssp. parviglumis; provided by UNAM, Cuernavaca, Mexico), one sorghum (Sorghum bicolor L. cv. Arprim; Semences de Provence, Fourques, France), one wheat (Triticum aestivum L. cv. Fiorina; provided by AgroScope, Changins, Switzerland) and one tomato (Solanum lycopersicum L. cv. Marmande; Vilmorin, La Ménitré, France) used as non-Poaceae, external reference.
We used the plant experiment carried out by Bouffaud et al.29. Plants were grown in loamy topsoil (sieved at 6 mm) collected in September 2009 from two adjacent fields (luvisols) located at La Côte Saint-André near Lyon (France). One is a maize-monocropping field (topsoil: clay 15.9%, silt 41.4%, sand 42.7%, organic matter 2.3%, pH (water) 7.3, N 1.6 g kg−1) and the other a permanent meadow (topsoil: clay 14.9%, silt 44.6%, sand 40.5%, organic matter 5.5%, pH (water) 6.0, N 3.2 g kg−1). Briefly, seeds were surface-disinfected and sown in 3-dm3 pots containing 2.5 kg soil (to obtain one seedling per pot) and the pots (including non-planted pots) were placed (randomized blocks; 5 pots per treatment) in a greenhouse. Sampling was carried out at 21 days in cropped soil for each treatment. In addition, maize lines FV4 and W85 (group Northern Flint) and tomato were also sampled at 21 days in meadow soil and at 42 days in cropped soil. Each root system was dug up, shaken vigorously (to eliminate soil loosely adhering to the roots), frozen (along with closely-adhering soil) in liquid nitrogen and lyophilized. Root and rhizosphere soil were separated and each stored at −20 °C (giving 0.1–0.7 g root samples and 0.5–6 g rhizosphere soil samples per plant). In addition, bulk soil was sampled in non-planted pots, at 21 (both soils) and 42 days, frozen, lyophilized and stored at −20 °C (giving 5 g samples).
Extraction of soil DNA and RNA
We used total nucleic acids that were extracted by Bouffaud et al.29, from 0.5 g of lyophilized rhizosphere or bulk soil using a protocol derived from Burgmann et al.44. Briefly, 0.5 g rhizosphere or bulk soil, 0.5 g zirconium beads (VWR, Fontenay-sous-Bois, France), 0.5 ml extraction buffer (5% hexadecyltrimethylammonium bromide, 1 mM 1,4-dithio-DL-threitol, in a 0.12 M phosphate buffer [pH 8]) were processed in a bead beater (TissueLyser II Retsch; Qiagen, Courtaboeuf, France) for 90 s at 30 m s−1. After 10 min centrifugation at 16,000 g, supernatants were extracted two times with phenol-chloroform-isoamyl alcohol (24:24:1 v/v/v) and then once with chloroform-isoamyl alcohol (24:1 v/v). Nucleic acids were precipitated overnight with potassium acetate (3 M, pH 4.8) and absolute ethanol at −20 °C. After centrifugation 30 min at 16,000 g, pellets were washed with 70% ethanol and dissolved in 100 μl RNase- and DNase-free water (giving 50–100 ng nucleic acids per μl).
cDNA synthesis by reverse transcription
Here, to obtain DNA-free RNA, 20 μl of nucleic acid solution were digested at room temperature with 4 U of DNase I (Invitrogen, Cergy Pontoise, France) in 1 × DNase I reaction buffer and RNA was purified using RNeasy Mini kit (Qiagen) following manufacturer’s instructions. Another step of DNA digestion was performed using the protocol described above to remove remaining traces of DNA and the reaction was stopped by incubating 10 min at 65 °C in presence of 1 μl of 25 mM EDTA.
Reverse transcription (RT) was performed on 8 μl of the resulting purified RNA extract using Omniscript reverse transcription kit (Qiagen; following the manufacturer’s instructions). The reaction was carried out 90 min at 37 °C. Inactivated was done at 95 °C (10 min) and cDNA was stored at −20 °C.
Quantitative PCR and quantitative RT-PCR
When selecting nifH primers, PolF/PolR9 were preferred over Zf/Zr31 because the latter proved ineffective for quantitative PCR (qPCR), giving non-specific products and smeared bands on gels4,9 and PolF/PolR is among recommended primers to capture nifH diversity efficiently19,20. Here, the amounts of nifH genes and transcripts (from rhizosphere or bulk soil) were estimated by qPCR and quantitative RT-PCR (qRT-PCR), after development of a real-time protocol based on the primers polF/polR9. The reaction was carried out in 20 μl containing 4 μl of PCR grade water, 2 μl of each primer (final concentration 0.50 μM), 10 μl of LightCycler-DNA Master SYBR Green I master mix (Roche Applied Science, Meylan, France) and 2 μl of sample DNA or cDNA (in triplicate for each of five repetitions per treatment). The cycling program included 10 min incubation at 95 °C, followed by 50 cycles of 95 °C for 15 s, 64 °C for 15 s and 72 °C for 10 s. Amplification specificity was studied by melting curve analysis of the PCR (and RT-PCR) products, performed by ramping the temperature to 95 °C for 10 s and back to 65 °C for 15 s, followed by increases of 0.1 °C s−1 up to 95 °C. Melting curve calculation and determination of Tm values were performed using the polynomial algorithm function of LightCycler Software v.1 (Roche Applied Science).
Genomic DNA from Azospirillum lipoferum 4B (whose genome contains one nifH copy) was used to generate standard curves, after dilution from 5 × 10−9 to 5 × 10−15 g DNA μl−1 (in triplicate). Sterile water was used as negative control for DNA amplification and DNAse treated RNA before reverse transcription was used as negative control for cDNA amplification. PCR efficiency was calculated from standard curves according to the equation E = 10(−1/slope). The five samples for each rhizosphere or bulk soil treatment were analyzed. Results in g μl−1 were converted in number of nifH copies using the following formula (assuming an average of 660 g mol−1 per base pair): number of copies = [DNA (g)× Avogadro’s number (molecules mol−1)]/[number of DNA base pairs in nifH fragment ×660 (g mol−1)]. The resulting numbers were expressed (i) per g of root, (ii) per root system and (iii) per g of soil.
T-RFLP analysis
For one replicate (individual plant or bulk soil sample) per treatment, nifH DNA and cDNA were amplified using forward primer polF9 5′-labeled with 6-FAM and reverse primer polR. PCR reaction was carried out in 50 μl containing 1 × buffer, 1 μM of each primer (Invitrogen), 2.5 mM of MgCl2, 1.75 U of Expand High Fidelity Taq polymerase (Roche Applied Science) and 2 μl of DNA or cDNA template. An initial denaturation at 94 °C for 2 min was followed by 30 cycles of 45 s denaturation at 94 °C, 30 s annealing at 55 °C and 30 s extension at 72 °C, followed by a final extension for 5 min. PCR products were purified using PCR purification kit (Macherey-Nagel, Hoerd, France). For T-RFLP, 500 ng of PCR product were digested using HaeIII (Fermentas, Villebon sur Yvette, France) 3 h at 37 °C and separated on automated sequencer ABI 3730XL (Applied Biosystems, Villebon sur Yvette, France). The number of individual terminal restriction fragments (T-RFs) was determined using GeneMapper v4.1 software (Applied Biosystems), with a detection limit of 50 relative fluorescence units.
Partial nifH sequencing
To corroborate T-RFLP data, qPCR products from DNA and cDNA samples of maize line W85 were cloned and sequenced, using the polF/polR primer set in 50 μl containing ~50 ng of purified qPCR products. PCR was carried out as described above. The PCR products were purified and cloned in the pGEMs-T Easy Vector System kit (Promega, Charbonnières, France) and positive clones were sequenced (Biofidal, Vaulx-en-Velin, France). Nucleotide sequences were analyzed using the SeaView multiplatform graphical user interface45 (available at http://pbil.univ-lyon1.fr/) using MUSCLE46 (default parameters) and phylogenetic trees inferred using PhyML46 (version 3.0) with a GTR model of nucleotide substitution47. Reference nifH genes were retrieved using BLASTP48 at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov).
Statistics
All analyses were done at P < 0.05, using R 2.10.1 software (http://www.r-project.org). First, treatments were compared concerning numbers of nifH genes and transcripts, using ANOVA and Fisher’s LSD tests, in each soil and at each sampling. Additionally, two-way ANOVA and Fisher’s LSD tests were performed to take into account past soil management at 21 days (i.e. treatment× past soil management) and sampling time effects in cropped soil (i.e. treatment × sampling + Error (samples/sampling).
Second, for T-RFLP data, treatments were compared based on peak presence/absence, using clustering analysis based on Euclidean distance and complete linkage clustering. One replicate (individual plant or bulk soil sample) was studied per treatment. For maize line W85, all five replicates were assessed, with similar results.
Third, Pearson correlation analysis was performed between (i) the phylogenetic distance between plant genotypes (Maximum Likelihood method, with the Kimura 2 parameter model, applied on three concatenated chloroplastic sequences i.e. gene rps16 and the intergenic spacers rps16-trnK and atpI-atpH29) and (ii) the corresponding pairwise differences in nifH numbers (genes or transcripts, expressed without log transformation), T-RFLP profiles (genes or transcripts), plant biomass (shoots or roots) or weight of root-associated soil (i.e. rhizosphere soil), based on the Euclidean distance.
Additional Information
How to cite this article: Bouffaud, M.-L. et al. Is plant evolutionary history impacting recruitment of diazotrophs and nifH expression in the rhizosphere? Sci. Rep. 6, 21690; doi: 10.1038/srep21690 (2016).
References
Herridge, D. F., Peoples, M. B. & Boddey, R. M. Global inputs of biological nitrogen fixation in agricultural systems. Plant Soil 311, 1–18 (2008).
Coelho, M. R. R. et al. Molecular detection and quantification of nifH gene sequences in the rhizosphere of sorghum (Sorghum bicolor) sown with two levels of nitrogen fertilizer. Appl. Soil Ecol. 42, 48–53 (2009).
Prakamhang, J., Minamisawa, K., Teamtaisong, K., Boonkerd, N. & Teaumroong, N. The communities of endophytic diazotrophic bacteria in cultivated rice (Oryza sativa L.). Appl. Soil Ecol. 42, 141–149 (2009).
Boyd, E. S. & Peters, J. W. New insights into the evolutionary history of biological nitrogen fixation. Front. Microbiol. 4 (2013).
Zehr, J. P., Jenkins, B. D., Short, S. M. & Steward, G. F. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ. Microbiol. 5, 539–554 (2003).
Wang, Y., Marschner, P. & Zhang, F. Phosphorus pools and other soil properties in the rhizosphere of wheat and legumes growing in three soils in monoculture or as a mixture of wheat and legume. Plant Soil 354, 283–298 (2011).
Pereira e Silva, M. C., Schloter-Hai, B., Schloter, M., van Elsas, J. D. & Salles, J. F. Temporal dynamics of abundance and composition of nitrogen-fixing communities across agricultural soils. PLoS ONE 8, e74500 (2013).
Collavino, M. M. et al. nifH pyrosequencing reveals the potential for location-specific soil chemistry to influence N2-fixing community dynamics. Environ. Microbiol. 16, 3211–3223 (2014).
Poly, F., Monrozier, L. J. & Bally, R. Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152, 95–103 (2001).
Wu, C. H., Bernard, S. M., Andersen, G. L. & Chen, W. Developing microbe–plant interactions for applications in plant-growth promotion and disease control, production of useful compounds, remediation and carbon sequestration. Microb. Biotechnol. 2, 428–440 (2009).
Junier, P., Junier, T., Witzel, K.-P. & Carú, M. Composition of diazotrophic bacterial assemblages in bean-planted soil compared to unplanted soil. Eur. J. Soil Biol. 45, 153–162 (2009).
Monteiro, J. M. et al. Bacterial communities within the rhizosphere and roots of vetiver (Chrysopogon zizanioides (L.) Roberty) sampled at different growth stages. Eur. J. Soil Biol. 47, 236–242 (2011).
Perin, L. et al. Diazotrophic Burkholderia species associated with field-grown maize and sugarcane. Appl. Environ. Microbiol. 72, 3103–3110 (2006).
Mao, Y., Yannarell, A. C., Davis, S. C. & Mackie, R. I. Impact of different bioenergy crops on N-cycling bacterial and archaeal communities in soil. Environ. Microbiol. 15, 928–942 (2013).
Mirza, B. S., Potisap, C., Nüsslein, K., Bohannan, B. J. M. & Rodrigues, J. L. M. Response of free-living nitrogen-fixing microorganisms to land use change in the Amazon rainforest. Appl. Environ. Microbiol. 80, 281–288 (2014).
Bulgarelli, D. et al. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403 (2015).
Wu, L., Ma, K. & Lu, Y. Prevalence of betaproteobacterial sequences in nifH gene pools associated with roots of modern rice cultivars. Microb. Ecol. 57, 58–68 (2009).
Knauth, S., Hurek, T., Brar, D. & Reinhold-Hurek, B. Influence of different Oryza cultivars on expression of nifH gene pools in roots of rice. Environ. Microbiol. 7, 1725–1733 (2005).
Wartiainen, I., Eriksson, T., Zheng, W. & Rasmussen, U. Variation in the active diazotrophic community in rice paddy-nifH PCR-DGGE analysis of rhizosphere and bulk soil. Appl. Soil Ecol. 39, 65–75 (2008).
Mårtensson, L. et al. Diazotrophic diversity, nifH gene expression and nitrogenase activity in a rice paddy field in Fujian, China. Plant Soil 325, 207–218 (2009).
Orr, C. H., James, A., Leifert, C., Cooper, J. M. & Cummings, S. P. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl. Environ. Microbiol. 77, 911–919 (2011).
de Dorlodot, S. et al. Root system architecture: opportunities and constraints for genetic improvement of crops. Trends Plant Sci. 12, 474–481 (2007).
Comas, L. H. & Eissenstat, D. M. Patterns in root trait variation among 25 co-existing North American forest species. New Phytol. 182, 919–928 (2009).
Czarnota, M. A., Rimando, A. M. & Weston, L. A. Evaluation of root exudates of seven sorghum accessions. J. Chem. Ecol. 29, 2073–2083 (2003).
Diamond, J. Evolution, consequences and future of plant and animal domestication. Nature 418, 700–707 (2002).
Osmont, K. S., Sibout, R. & Hardtke, C. S. Hidden branches: Developments in root system architecture. Annu. Rev. Plant Biol. 58, 93–113 (2007).
Uga, Y. et al. Variation in root morphology and anatomy among accessions of cultivated rice (Oryza sativa L.) with different genetic backgrounds. Breed. Sci. 59, 87–93 (2009).
Lambert, J. B., Heckenbach, E. A., Wu, Y. & Santiago-Blay, J. A. Characterization of plant exudates by principal-component and cluster analyses with nuclear magnetic resonance variables. J. Nat. Prod. 73, 1643–1648 (2010).
Bouffaud, M.-L., Poirier, M.-A., Muller, D. & Moënne-Loccoz, Y. Root microbiome relates to plant host evolution in maize and other Poaceae. Environ. Microbiol. 16, 2804–2814 (2014).
Penton, C. R. et al. Functional genes to assess nitrogen cycling and aromatic hydrocarbon degradation: primers and processing matter. Front. Microbiol. 4 (2013).
Zehr, J. P. & McReynolds, L. A. Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl. Environ. Microbiol. 55, 2522–2526 (1989).
Gaby, J. C. & Buckley, D. H. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS ONE 7, e42149 (2012).
Messer, L. F. et al. High levels of heterogeneity in diazotroph diversity and activity within a putative hotspot for marine nitrogen fixation. ISME J. (2015), doi: 10.1038/ismej.2015.205.
Provorov, N. A. & Vorobyov, N. I. Host plant as an organizer of microbial evolution in the beneficial symbioses. Phytochem. Rev. 8, 519–534 (2009).
Parker, M. A. & Spoerke, J. M. Geographic structure of lineage associations in a plant-bacterial mutualism. J. Evol. Biol. 11, 549–562 (1998).
Lugtenberg, B. & Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 63, 541–556 (2009).
Raaijmakers, J. M., Paulitz, T. C., Steinberg, C., Alabouvette, C. & Moënne-Loccoz, Y. The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 321, 341–361 (2008).
Vacheron, J. et al. Plant growth-promoting rhizobacteria and root system functioning. Front. Plant Sci. 4 (2013).
Mehnaz, S., Kowalik, T., Reynolds, B. & Lazarovits, G. Growth promoting effects of corn (Zea mays) bacterial isolates under greenhouse and field conditions. Soil Biol. Biochem. 42, 1848–1856 (2010).
Meyer, J. B. et al. Interplay between wheat cultivars, biocontrol pseudomonads and soil. Appl. Environ. Microbiol. 76, 6196–6204 (2010).
Foster, R. A., Subramaniam, A. & Zehr, J. P. Distribution and activity of diazotrophs in the Eastern Equatorial Atlantic. Environ. Microbiol. 11, 741–750 (2009).
Bannert, A. et al. Changes in diversity and functional gene abundances of microbial communities involved in nitrogen fixation, nitrification and denitrification in a tidal wetland versus paddy soils cultivated for different time periods. Appl. Environ. Microbiol. 77, 6109–6116 (2011).
Dandie, C. E. et al. Abundance, diversity and functional gene expression of denitrifier communities in adjacent riparian and agricultural zones. FEMS Microbiol. Ecol. 77, 69–82 (2011).
Bürgmann, H., Widmer, F., Sigler, W. V. & Zeyer, J. mRNA extraction and reverse transcription-PCR protocol for detection of nifH gene expression by Azotobacter vinelandii in soil. Appl. Environ. Microbiol. 69, 1928–1935 (2003).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 (2010).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Acknowledgements
This work was supported in part by the Ministère Français de la Recherche. We are grateful to J. Vacheron and I. Jacquemond for technical assistance and D. Abrouk (iBio platform, UMR CNRS 5557 Ecologie Microbienne) for helpful discussion.
Author information
Authors and Affiliations
Contributions
M.L.B., Y.M.L. and D.M. conceived and designed the project. M.L.B. performed green house experiment and molecular biology experiments and the statistical analysis. S.R. analyzed nifH sequences. M.L.B., Y.M.L. and D.M. analyzed the findings and wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bouffaud, ML., Renoud, S., Moënne-Loccoz, Y. et al. Is plant evolutionary history impacting recruitment of diazotrophs and nifH expression in the rhizosphere?. Sci Rep 6, 21690 (2016). https://doi.org/10.1038/srep21690
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21690
This article is cited by
-
Contribution of soil diazotrophs to crop nitrogen utilization in an acidic soil as affected by organic and inorganic amendments
Plant and Soil (2024)
-
Promotion of biological nitrogen fixation activity of an anaerobic consortium using humin as an extracellular electron mediator
Scientific Reports (2021)
-
How do different nitrogen application levels and irrigation practices impact biological nitrogen fixation and its distribution in paddy system?
Plant and Soil (2021)
-
Diazotrophic communities are more responsive to maize cultivation than phosphorus fertilization in an acidic soil
Plant and Soil (2020)
-
1-Aminocyclopropane-1-carboxylate deaminase producers associated to maize and other Poaceae species
Microbiome (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
