
Abstract
Abnormal level of Serum Uric Acid (SUA) is an important marker and risk factor for complex diseases including Type 2 Diabetes. Since genetic determinant of uric acid in Indians is totally unexplored, we tried to identify common variants associated with SUA in Indians using Genome Wide Association Study (GWAS). Association of five known variants in SLC2A9 and SLC22A11 genes with SUA level in 4,834 normoglycemics (1,109 in discovery and 3,725 in validation phase) was revealed with different effect size in Indians compared to other major ethnic population of the world. Combined analysis of 1,077 T2DM subjects (772 in discovery and 305 in validation phase) and normoglycemics revealed additional GWAS signal in ABCG2 gene. Differences in effect sizes of ABCG2 and SLC2A9 gene variants were observed between normoglycemics and T2DM patients. We identified two novel variants near long non-coding RNA genes AL356739.1 and AC064865.1 with nearly genome wide significance level. Meta-analysis and in silico replication in 11,745 individuals from AUSTWIN consortium improved association for rs12206002 in AL356739.1 gene to sub-genome wide association level. Our results extends association of SLC2A9, SLC22A11 and ABCG2 genes with SUA level in Indians and enrich the assemblages of evidence for SUA level and T2DM interrelationship.
Similar content being viewed by others
Introduction
Uric acid is a by-product of oxidation of purine. SUA levels have been used as biological marker for many disorders like gout, arthritis, kidney functions1,2, hypertension, metabolic disorders and type 2 diabetes3,4. Studies have established SUA as an important stake holder regarding health issues of particular population. Hence it creates a necessity to study factors affecting SUA level of a population.
The levels of uric acid in an individual is a combined result of genetic factors and multitude of life style related factors like food habit, exercise, work type and means of transportation5,6,7,8. Indians differ in their food habit, living style and genetic constitutions from other ethnic populations in the world9,10,11.
Genetic studies have established a heritability of 40–70% for SUA level suggesting stronger role of genetic factors in determining SUA level12. Major part of genetic factors contributing to the SUA level has not been well understood as few number of genetic studies have been performed in limited populations and most of them being of European ethnicity. GWAS conducted on Japanese, Chinese, African American and Amish populations have established association of loci in urate transporter genes like SLC2A9, ABCG2, SLC22A1 and SLC22A1213,14,15,16,17,18,19. Large scale meta-analysis conducted by Asian Genetic Epidemiology Network (AGEN) has included some samples from the Singapore Indian Study (SINDI) and identified loci in transcription factor MAF for SUA level20. Another large scale meta-analysis conducted by Global Urate Genetics Consortium15 identified 18 new loci associated with SUA level near TRIM46, INHBB, SFMBT1, TMEM171, VEGFA, BAZ1B, PRKAG2, STC1, HNF4G, A1CF, ATXN2, UBE2Q2, IGF1R, NFAT5, MAF, HLF, ACVR1B-ACVRL1 and B3GNT4 genes. Their analysis in 8380 samples of Indian ancestry showed association of variants in SLC2A9, ABCG2, SLC22A11, GCKR, SLC17A1 gene at genome wide significance level. None of these study subjects were living in India. Both of the studies include Indian subjects from different ethnicities including Dravidian samples. This reflects a lack of independent studies conducted on Indian subjects for SUA level globally despite several waves of GWAS for different phenotype in different population. There is no separate genetic epidemiological study for uric acid levels in Indian population till date. Hence, genetic study in Indian population becomes necessary and provides a unique opportunity to explore the population specific genetic factors affecting uric acid level related to Indians.
Some of the identified genetic loci associated with SUA levels are found to show interactions with several phenotypes like sex, age and Body Mass Index (BMI)21,22. A recent epidemiological study showed a stronger association of SUA level with impaired fasting glucose suggesting a complex relationship between uric acid pathophysiology and glucose level23. T2DM is a condition where glucose metabolism of the individual gets impaired along with several other biochemical and signaling pathways. Another report showed association of uric acid transporter gene SLC2A9 variant rs1014290 with T2DM status24. This suggests a plausible inter-relation between SUA related genetic factors and T2DM. However, there is no concrete study till date investigating the interaction between SUA genetics and T2DM. The present study aims at identification of common variants associated with SUA levels by two-staged Genome Wide Association Study (GWAS) in 4,834 healthy Indians of Indo-European ethnicity living in Northern part of India. Further, it extends its interest to explore the variability in effect of identified GWAS variants under altered condition (T2DM).
Results
Genome wide association analysis of SUA
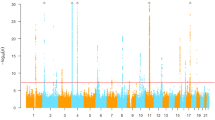
After stringent quality control, we analyzed a total of 5,39,662 genetic markers in discovery phase for their association with SUA levels in 1,109 individuals using linear regression. A good agreement was observed between the theoretical p-value distribution and calculated p-values using QQ plot as shown in Fig. 1. The genomic inflation factor for the fitted model was calculated as 1.006 that indicates homogeneity of analyzed samples. SPATA13 variants (rs9511097) were detected as most significant signal (Effect size = −16.35, p-value = 2.19 × 10−6) in discovery phase (Fig. 2). Along with the earlier known GWAS signals for SUA, SNPs with p-values<10−4 from discovery phase were genotyped in 3,725 Indian samples for replication phase. 4 SNPs associated with SUA level in 1st phase had to be removed before final analysis in replication phase owing to their failure in quality control.

The -log10 of p-values observed for the association of SNPs in discovery phase genome wide association analysis under additive model adjusted for age, sex BMI, PC1 and PC2 (black symbols) are plotted against the theoretical -log10 p-values expected under the null hypothesis (red line). The genomic control inflation factor (λ) was estimated to be 1.06.

The -log10 p-values for association of genotyped SNPs are plotted as a function of genomic position (National Center for Biotechnology Information Build 37). The p-values were determined using logistic regression adjusted for age, sex, BMI, PC1 and PC2 in discovery phase analysis. Each chromosome (Chr) has been represented with a unique color.
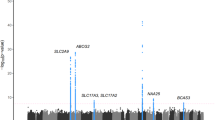
Meta-analysis was performed in 4,834 normoglycemic individuals and results yielded five SNPs in two different genes (SLC2A9 and SLC22A11) associated with SUA levels at genome wide significance levels (Table 1, Fig. 2). Variant rs3775948 in SLC2A9 gene showed most significant association with SUA levels in Indians (p-value = 1.7 × 10−19) that is in line with earlier findings in Japanese and African American population (Table 2)13,14. Another three detected variants in SLC2A9 gene include rs16890979 (p-value = 2.62 × 10−18), rs11722228 (p-value = 7.34 × 10−16) and rs737267 (p-value = 2.71 × 10−16). The other genetic variant (rs2078267) attaining genome wide significant level was found to reside in SLC22A11 gene (p-value = 3.26 × 10−11) (Table 1, Fig. 2). We also noticed almost genome wide association for missense variant rs2231142 (p-value = 7.82 × 10−8) in ABCG2 gene (Table 1).
Conditional analysis to examine independence of signals in SLC2A9
To examine independent association of signals in SLC2A9, conditional analysis of cumulative data (discovery and replication phase) was performed using additive model and result showed that rs3775948 (SLC2A9) as the lead SNP (Supplementary Table S1). Following conditional analysis for rs3775948, none of remaining three SNPs showed association at genome wide significance level but remained significant at sub-GWAS level (rs16890979, p-value = 1.078 × 10−7; rs737267, p-value = 1.86 × 10−5 and rs11722228, p-value = 1.64 × 10−5) (Supplementary Table S1). Results also indicated moderate linkage disequilibrium between rs3775948 and rs16890979 (R2 = 0.5).
Status of earlier known GWAS signals
To examine the status of SUA associated variants across various populations, we compared effect size and association strength of known signals reported in GWAS catalogue among Indians and other major ethnic populations. Variants in SLC22A12 gene (rs505802, P = 5.18 × 10−6) and GCKR gene (rs1260326, P=7.7 × 10−4 and rs780094, P=5.83 × 10−3) achieve sub-genome wide significance level in Indian population (Table 2). In addition, nominal association of variant rs1165205 in SLC17A3 gene (P = 0.03) with SUA level was detected in studied samples, whereas, markers in PDZK1 (rs1967017), ATXN2 (rs653178), SLC17A1 (rs1165196), BTF3P7-RREB (rs675209) and SLC16A9 (rs12356193) could not be replicated (Table 2).
Furthermore, difference in the effect size of genetic variants was also observed across population. Variants rs3775948 (SLC2A9, G allele) has been associated with decrease of uric acid level in Japanese population (Table 2) and African American population14, however it has higher effect size in Indians. Another non-synonymous coding SNP rs16890979 (A allele-Val253Ile) in exon 8 of SLC2A9 gene has been associated with decrease in SUA levels in Chinese, European and Amish population17,18,19. The effect size of this variant in Indians is less than Caucasian and Amish population and higher than Chinese population (Table 2). The variant rs2078267 (A allele) in SLC22A11 gene has higher effect size in Indians than in Caucasian population. Furthermore, ABCG2 missense variant (rs2231142, Gln141Lys) has higher effect size in Indians than Caucasian, Chinese and Japanese population as shown in Table 2.
Increase in number of GWAS signals with advent of type 2 diabetes
The effect of increasing sample size on the status of SUA associated genetic variants was examined. For this, meta-analysis of summary statistics obtained from association of T2DM subjects was conducted along with summary statistics from normoglycemic subjects. Analysis in a total sample of 5,911 individuals consisting of 4,834 normoglycemic subjects and 1,077 T2DM subjects improved the association of missense variant rs2231142 in ABCG2 from 7.82 × 10−8 (Table 1) in normoglycemic subjects to 1.31 × 10−10 in combined sample (Table 3). Inclusion of T2DM samples in combined meta-analysis did not affect homogeneity of association (Table 3) and no difference in minor allele frequency (MAF) of rs2231142 was seen in normoglycemic and T2DM samples (Table 4).
SUA variants have higher effect size in T2DM subjects
To unravel the possible inter-relation of SUA associated genetic variants with type 2 diabetes, we sought to examine whether these signals have different fate and effects in T2DM patients. We compared the effect sizes of signals that achieved GWAS level significance and were also significantly associated (p-value = 0.05) with SUA in T2DM subjects. Analysis revealed significant differences in the effect size of these variants between T2DM patients and normoglycemic subjects. Variants rs16890979 (A allele, d = −2.74) and rs737267 (A allele, d = −3.26) in SLC2A9 genes possess higher effect size in presence of T2DM. Additionally, ABCG2 gene variant rs2231142 (A allele, d = 1.71) was also reported to be associated with high SUA levels in T2DM patients (Table 4). Therefore, higher effect sizes of SUA genetic determinants in T2DM suggest possible interaction with T2DM pathophysiology. Joint analysis including T2DM patients and normoglycemic subjects adjusted for age, sex, BMI and diabetic status also confirmed the association of above six variants with SUA level at genome wide significance in Indian population. It also confirmed association of rs2231142 variant in ABCG2 gene with SUA level in Indians as shown in Table 5.
Genotypes based on individual SNPs strongly correlate with average SUA level of subjects in Indians as shown in Supplementary Fig. S2. The cumulative effect analysis also revealed strong association of genetic score with SUA level (p-value = 2 × 10−16). Each additional allele causes a 6.45 unit increase in SUA level as shown in Supplementary Fig. S3.
Enrichment analysis of the significant genes
Enrichment analysis was performed for the genes found significant in meta-analysis and result showed urate metabolic process as significantly enriched process in Indian population (Supplementary Table S2). Enrichment of six genes (GCKR, SLC2A9, ABCG2, SLC22A11, SLC22A12 and SLC17A3) were observed against 11 genes (PRPS1, SLC17A1, SLC22A11, SLC2A9, PNP, SLC22A12, SLC16A9, GCKR, SLC17A3, ABCG2 and LRC16A) present in human genome under urate metabolic process (accession number GO:0046415) with IMP (Inferred from mutant phenotype) category.
Identification of novel signals in healthy subjects
Although our analysis in healthy subjects could not detect any novel genetic variants associated with SUA levels at genome wide significance levels, we have identified three novel loci (rs12206002, rs993701, rs1445305) in two different genes (UTRN-AL356739.1, AC064865.1-RPL6P5) at sub-genome wide significance levels (p-value<10−5, 10−4) (Supplementary Table S3). These loci could not be detected at genome wide levels even when our study was sufficiently powered (>98%) to detect these associations (Supplementary Fig. S1).
Meta-analysis and in-silico replication
The meta-analysis of summary statistics for three novel loci (rs12206002, rs993701 and rs1445305) was performed with association data from AUSTWIN consortium. Meta-analysis improved the association status for variant rs12206002 from 8.83 × 10−5 to 7.23 × 10−7 (Supplementary Table S4). Heterogeneity in effect size of rs12206002 was observed across populations. We did not detect any improvement in the association of variant rs993701as it shifted to a meta-analysis p-value of 0.45 from 1.23 × 10−4 in Indian normoglycemic subjects. Association p-value for variant rs1445305 also increased from 9.85 × 10−4 to 0.21 after meta-analysis.
Discussion
Present study is the first genome wide association study for SUA level in Indians. We found association of SLC2A9, SLC22A11 and ABCG2 gene variants at genome wide significance level (p-value < 10−8). This is the first study to report the differences in effect size of SUA associated genetic variants in SLC2A9 and ABCG2 in T2DM patients that suggests involvement of these gene variants in the alteration of uric acid levels in T2DM subjects. We also replicated variants in GCKR and SLC17A1 gene at nominal significance levels. All these genes encode for transporter proteins. All significant genes explained nearly ~6% of the variance explained in uric acid level. This suggests contribution of other genetic, epigenetic and other factors in pathophysiology of uric acid levels in Indian population.
Studies have reported that SLC2A9 is expressed in both kidney and liver of human and mice and is upregulated in diabetes mice25. The SLC2A9 expression was found to be governed by p53 gene and is mediated by oxidative stress26. Oxidative stress play major and deterministic role in patho-physiology of T2DM and has been observed to be higher in T2DM patients than healthy controls27. The higher expression of SLC2A9 in diabetic condition may be governed by higher oxidative stress in diabetics. In a recent study, Hurba et al. observed that there is no significant difference in transport activity of coding rs16890979 (Val253Ile) variant containing protein and wild type protein in Xenopus oocyte expression system28. The higher activity of SLC2A9 in T2DM subjects compared to normoglycemics may be attributed to higher expression of total SLC2A9 protein in T2DM condition.
Similarly, another significant gene ABCG2 is located at apical membrane of renal proximal tubule and is involved in excretion of uric acid in urine. The functional form of ABCG2 protein present at cell membrane is homodimer glycosylated protein29,30. The homodimer formation requires proper folding and a disulphide bond formation between residues of two monomers in endoplasmic reticulum29. Reducing folding environment has found to induce improper folding in Q141K containing protein31. T2DM is a complex disorder where many biochemical processes, signaling cascades as well as hormone levels get altered. One of the major consequences of T2DM is alteration in redox potential in cellular compartments as well as extracellular spaces32. Study suggests that although there is an increase in oxidative environment of other compartments, the redox potential of microsomal vesicles from diabetic mice was found to shift towards more reducing end32. Because of these alterations of oxidative environment of microsomal vesicles towards comparatively more reducing environment in diabetic patients, more of the Q141K containing protein will be improperly folded and will be prone for proteosomal degradation. This will cause lesser protein trafficking, glycosylation and presence at the cell surface, finally lesser activity. This decrease in activity may increase the SUA level more in T2DM subjects than normoglycemics (Fig. 3). So far by our knowledge, there is no study to show evidence for T2DM interaction with SLC2A9 and ABCG2 genotypes. The observation regarding modulation in effect size of SUA associated variants in T2DM patients need to be confirmed in larger sample size and across different population. Possibility of any alternative mechanisms that modulates the effect of variants in SLC2A9 and ABCG2 genes under diabetic condition cannot be excluded.

Mode of action of variants in ABCG2 and SLC2A9 genes under T2DM condition.
Our study also showed the association of rs2078267 variant in SLC22A11 gene at genome wide significance level in any population outside Caucasians for the first time. SLC22A11 is a low affinity urate transporter and known to express in kidneys and placenta. SLC22A11 has a role in the transport of multiple organic anions33.
Allele dosage analysis suggests that accumulation of studied variants in number may heavily contribute towards increasing SUA level in Indians.
Our study identified three novel variants near two different long non-coding RNA genes (AL356739.1, AC064865.1) at sub-genome wide levels (p-value < 10−5, 10−4) (Supplementary Table S3) in normoglycemics subjects. Stronger association of variant rs12206002 suggests its indispensable role to play in pathophysiology of SUA level in Indians and various other populations. Novel variants rs993701 and rs12206002 are 123kb and 130kb upstream of AL356739.1 gene. Variant rs1445305 is 4.7 kb downstream of AC064865.1 gene. Functions of these genes are only speculative and not confirmed. The variant rs1445305 is on an enhancer mark in K562 cell line. This suggests a possible functional role of these variants by altering the binding of transcription related protein.
In conclusion, our study extended the association of three uric acid transporter genes with SUA level to Indians at genome-wide significant level. It also indicated alteration in effect size of the genetic variants associated with SUA level with disease condition like T2DM. Our study further suggests involvement of both common as well as population specific genetic player to determine SUA levels. Enrichment of urate metabolic process in significant genes represents the contribution of uric acid transporter genes in determining SUA levels in Indians. These observations may have implications for further research into genetics of urate signaling in Indian population. These findings will also have possible impacts in pharmaceutical industry to understand the efficacy of urate lowering drug in altered condition like T2DM. The current information may also find relevance during treatment of patients with T2DM along with SUA related complications and may help in their better management.
Methodology
Study participants
All study participants in this study are the member of INdian DIabetes Consortium34. Samples are well characterized for anthropometric and biochemical parameters34 (Supplementary Table S5). These samples were enrolled in the study by conducting diabetes awareness camp organized in various parts of North India. Prior informed written consent was obtained from the study participants. The study was approved by the Human Ethics Committee of CSIR-Institute of Genomics and Integrative Biology and All India Institute of Medical Sciences research Ethics Committee. The study was conducted in accordance with the principles of Helsinki Declaration. Various anthropometric parameters like height, weight, waist circumference, hip circumference and biochemical parameters like total cholesterol, triglyceride, HDL and LDL were measured. The uric acid level was measured by enzymatic colorimetric method using COBAS Integra 400 plus (Roche Diagnostic, Mannheim, Germany).
Discovery phase genotyping and quality control
Discovery phase samples were genotyped as a part of T2DM GWAS conducted in our laboratory using Illumina Human 610-quad bead chips11. Normoglycemic individuals used as control for T2DM GWAS were analyzed for current study. Genotype of these samples has been called by Gene call algorithm using Genome studio software. Strong quality control (both samples and SNPs) were performed on data. Samples were excluded based on call rate (<95%), heterozygosity (Samples with observed heterozygosity value 3 SD away from the mean heterozygosity were removed) and sex-discrepancy. Related samples were removed based on Identity-by-descent analysis (Pi-hat>0.1875). Cryptic relatedness for samples was calculated using 1,17,982 pruned SNPs. Pruning of the SNPs was done by applying –indep-pairwise command with a r2 0.2 and window size of 50 using PLINK v1.07 (http://pngu.mgh.harvard.edu/~purcell/plink)35. Population outliers were detected using principal component analysis (http://www.complextraitgenomics.com/software/gcta/)36. First five principal components were used to detect population outlier samples. A total of 27 samples having eigenvectors 6 standard deviation away from the mean value were removed as outliers. SNPs with call rate (<97%), Hardy Weinberg equilibrium (p-value < 10−5) and MAF <0.01 were removed. We also removed SNPs in sex and mitochondrial chromosomes. Details for quality control steps have been given as Supplementary Fig. S4.
Samples with SUA level 3 standard deviations away of mean value were considered as outliers and were removed from the analysis. SUA levels were converted in to standard unit (μmol/L) and then inverse normalized using inbuilt command in R (http://www.r-project.org/). Association of SNPs with inverse normalized SUA levels was tested using linear regression model in PLINK. Sex, age, BMI and first three principal components were used as covariates in the model. To find the deviation of p-values obtained from an additive model a quantile-quantile (QQ) plot for –log (p-value) against theoretical p-value was plotted using qqman package in R.
Validation phase genotyping and meta-analysis
Genetic markers from discovery phase having p-value < 10−4, in addition to known signals associated with SUA levels were taken for validation phase. The selected markers were genotyped along with other markers selected for ongoing GWAS for other quantitative traits in our laboratory using GoldenGate technology (Illumina San Diago, USA) and were analyzed in 3,725 Indians. A total of 204 samples (5.47%) were genotyped in duplicate, error rate of <0.01 was detected. Samples were removed having call rate <90%. SNPs with GenTran score <0.60, cluster separation score <0.4 and call rate <90% were excluded. We also excluded SNPs with minor allele frequency <0.01 and Hardy-Weinberg Equilibrium p-value <10−5. Samples with SUA level 3 standard deviations away from mean value were removed. SUA level was converted to standard unit and was inverse normalized before association. Association analysis was performed using linear regression model and model was adjusted for age, sex and BMI. Conditional analysis for loci in SLC2A9 was done in the merged healthy samples using PLINK and model was adjusted for age, sex and BMI as covariates. Meta-analysis of the summary statistics of the stage 1 and stage 2 results was done by METAL (ttp:// www.sph.umich.edu/csg/abecasis/Metal/) using fixed effect inverse variance method37. All analyses have been done in healthy subjects if not stated otherwise.
We have also done joint analysis of both discovery phase and replication phase normoglycemic and T2DM subjects in seeking for association of some additional signals with SUA level. Joint analysis was done using linear regression and model was adjusted for age, sex, BMI and T2DM status.
Enrichment analysis of the significant genes
To identify the functional contribution of the genes significantly associated with SUA levels, enrichment analysis was performed using GeneMANIA (http://www.genemania.org/)38. Enrichment for function of significant genes (p-value < 0.05) obtained during meta-analysis of normoglycemic subjects was done by searching for functions in GeneMANIA with pathway as network. Features with FDR corrected p-value < 0.05 were considered significant.
In-silico Analysis of effect size of SUA associated variants in European ancestry subjects
The top three novel signals obtained from meta-analysis of stage 1 and stage 2 results in Indians were tested for association with SUA level using genome wide data from Australian Twin-Family Study (AUSTWIN) consortium in Australian samples of European origin39. AUSTWIN now includes 11741 adult participants (twins and their family members) with genotyping and uric acid results. Meta-analysis by combining the summary statistics for association in normolycemic subjects, T2DM subjects and AUSTWIN subjects was done by METAL. The effect sizes were converted to uniform unit by proper conversion factor before meta-analysis.
Analysis of effect size of SUA associated variants in T2DM subjects
To determine possible difference in the impact of SUA associated genetic variants in diabetic conditions, we examined the effect size of markers associated with uric acid level at genome wide significance level in 1,077 T2DM subjects. We selected only those markers that were associated with SUA level (p-value<0.05) in both T2DM subjects and normoglycemic subjects. Data were analyzed separately for T2DM similar to normoglycemic subjects and effect size was estimated as mentioned above. Cohen’s d (d) value was calculated to compare the effect size. Cohen’s d was calculated as the ratio of difference of effect size between T2DM and normoglycemic divided by pooled variance of the two effect sizes.
Combined risk score analysis
To find the cumulative affect of SUA increasing alleles for studied GWAS variant, effective unweighted genetic risk score was calculated. Genotypes were coded as 0, 1 and 2 based upon presence of zero, single or double SUA increasing allele in a subject. The effective genetic score was calculated as sum of scores for all six variants. To study the comparative enhancement in SUA level, subjects were divided into <2, 2–3, 4–5, 6–7, 8–9 and 10–11 genetic score containing groups. Mean value of SUA level was calculated and plotted against genetic score.
Power calculation
Power of the study was calculated using Quanto software (http://hydra.usc.edu/gxe/) assuming additive genetic model for a range of allele frequencies from 0.001–0.5. Two tailed test at significance level of 0.05 with effect size ranging from 10.50–16.50 obtained from literature was used for power calculation and it was found that present study is sufficiently powered to detect association of genetic variants. A urate level of 308 μmol/L and standard deviation of 91 μmol/L was used. MAF for combined samples and calculated effect size from meta-analysis was used for final power analysis.
Additional Information
How to cite this article: Giri, A. K. et al. Genome wide association study of uric acid in Indian population and interaction of identified variants with Type 2 diabetes. Sci. Rep. 6, 21440; doi: 10.1038/srep21440 (2016).
References
Gibson, T. Hyperuricemia, gout and the kidney. Curr. Opin. Rheumatol. 24, 127–131 (2012).
Perez-Ruiz, F., Calabozo, M., Erauskin, G. G., Ruibal, A. & Herrero-Beites, A. M. Renal underexcretion of uric acid is present in patients with apparent high urinary uric acid output. Arthritis Rheum. 47, 610–613 (2002).
Dehghan, A., Hoek, M. Van, Sijbrands, E. J. G., Hofman, A. & Wetteman, J. C. M. High Serum Uric Acid as a Novel Risk Factor for Type 2 Diabetes. Cardiovasc. Metab. Risk. 31(2), 361–362 (2008).
Sluijs, I. et al. Plasma Uric Acid Is Associated with Increased Risk of Type 2 Diabetes Independent of Diet and Metabolic Risk Factors. J. Nutr. 143, 80–85 (2013).
Huang, L. L., Huang, C. T., Chen, M. L. & Mao, I. F. Effects of profuse sweating induced by exercise on urinary uric acid excretion in a hot environment. Chin. J. Physiol. 53, 254–261 (2010).
Green, H. J. & Fraser, I. G. Differential effects of exercise intensity on serum uric acid concentration. Med. Sci. Sports Exerc. 20, 55–59 (1988).
Schmidt, J. A., Crowe, F. L., Appleby, P. N., Key, T. J. & Travis, R. C. Serum Uric Acid Concentrations in Meat Eaters, Fish Eaters, Vegetarians and Vegans: A Cross-Sectional Analysis in the EPIC-Oxford Cohort. PLoS One 8(2), e56339 (2013).
Liu, L. et al. Relationship between lifestyle choices and hyperuricemia in Chinese men and women. Clin. Rheumatol. 32, 233–239 (2013).
Giri, A. K. et al. Pharmacogenetic landscape of clopidogrel in north Indians suggest distinct interpopulation differences in allele frequencies Pharmacogenomics. 15(5), 643–653 (2014).
Tabassum, R. et al. No association of TNFRSF1B variants with type 2 diabetes in Indians of Indo-European origin. BMC Med. Genet. 12, 110 (2011).
Tabassum, R. et al. Genome-wide association study for type 2 diabetes in indians identifies a new susceptibility locus at 2q21. Diabetes 62, 977–986 (2013).
Nath, S. D. et al. Genome scan for determinants of serum uric acid variability. J. Am. Soc. Nephrol. 18, 3156–3163 (2007).
Kamatani, Y. et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 42, 210–216 (2010).
Charles, B. A et al. A genome-wide association study of serum uric acid in African Americans. BMC Med. Genomics 4, 17 (2011).
Köttgen, A. et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 45, 145–154 (2013).
Yang, Q. et al. Multiple Genetic Loci Influence Serum Urate and Their Relationship with Gout and Cardiovascular Disease Risk Factors. Circ. Cardiovasc. Genet. 3, 523–530 (2010).
Yang, B. et al. A genome-wide association study identifies common variants influencing serum uric acid concentrations in a Chinese population. BMC Med. Genomics 7, 10 (2014).
Dehghan, A. et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372, 1953–1961 (2008).
McArdle, P. F. et al. A common nonsynonymous variant in GLUT9 is a determinant of serum uric acid levels in old order Amish. Arthritis Rheum. 58, 2874–2881 (2008).
Okada, Y. et al. Meta-analysis identifies multiple loci associated with kidney function–related traits in east Asian populations. Nat. Genet. 44, 904–910 (2012).
Brandstatter, A. et al. Sex-specific association of the putative fructose transporter SLC2A9 variants with uric acid levels is modified by BMI. Diabetes Care. 31, 1662–1667 (2008).
Brandstätter, A. et al. Sex and age interaction with genetic association of atherogenic uric acid concentrations. Atherosclerosis 210, 474–478 (2010).
Kawamoto, R. et al. Serum Uric Acid Is More Strongly Associated with Impaired Fasting Glucose in Women than in Men from a Community-Dwelling Population. PLoS One 8, 1–5 (2013).
Liu, W. C. et al. The rs1014290 polymorphism of the SLC2A9 gene is associated with type 2 diabetes mellitus in Han Chinese. Exp. Diabetes Res. 2011, Article ID 527520 (2011).
Keembiyehetty, C. et al. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are up-regulated in diabetes. Mol. Endocrinol. 20, 686–697 (2006).
Itahana, Y. et al. The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene 34, 1799–1810 (2015).
Folli, F. et al. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Curr. Diabetes Rev. 7(5), 313–324 (2011).
Hurba, O. et al. Complex Analysis of Urate Transporters SLC2A9, SLC22A12 and Functional Characterization of Non-Synonymous Allelic Variants of GLUT9 in the Czech Population: No Evidence of Effect on Hyperuricemia and Gout. PLoS One 9, e107902 (2014).
Age, K. K. et al. Dominant-Negative Inhibition of Breast Cancer Resistance Protein as Drug Efflux Pump through the Inhibition of S-S Dependent Homodimerization. Int. J. Cancer 97, 626–630 (2002).
Diop, N. K. & Hrycyna, C. A. N-linked glycosylation of the human ABC transporter ABCG2 on asparagine 596 is not essential for expression, transport activity, or trafficking to the plasma membrane. Biochemistry 44, 5420–5429 (2005).
Litman, T. et al. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim. Biophys. Acta. 1565, 6–16 (2002).
Nardai, G., Korcsmáros, T., Papp, E. & Csermely, P. Reduction of the endoplasmic reticulum accompanies the oxidative damage of diabetes mellitus. Biofactors 17, 259–267 (2003).
Zhou, F., Zhu, L., Cui, P. H., Church, W. B. & Murray, M. Functional characterization of nonsynonymous single nucleotide polymorphisms in the human organic anion transporter 4 (hOAT4). Br. J. Pharmacol. 159, 419–427 (2010).
INdian DIabetes COnsortium. INDICO: the development of a resource for epigenomic study of Indians undergoing socioeconomic transition. The HUGO journal. 5, 65–69 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Montojo, J. et al. GeneMANIA cytoscape plugin: Fast gene function predictions on the desktop. Bioinformatics 26, 2927–2928 (2010).
Middelberg, R. P. S. et al. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC Med. Genet. 12, 123 (2011).
Kolz, M. et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 5(6), e1000504 (2009).
Vitart, V. et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 40, 437–442 (2008).
Acknowledgements
The authors are thankful to all the participating subjects for their support and cooperation in carrying out the study. The authors acknowledge the support and participation of members of the INDICO consortium in the generation of data. We also thank to AUSTWIN consortium for providing the summary statistics for meta-analysis. We heartily thank Dr. Abhay Sharma for his critical evaluation of the manuscript. This work was supported by the Council of Scientific and Industrial Research [CSIR], Government of India through Centre for Cardiovascular and Metabolic Disease Research [CARDIOMED] project [Grant No: BSC0122-(7)].
Author information
Authors and Affiliations
Contributions
A.K.G. assembled and analyzed the data; contributed to the discussions and wrote the manuscript. P.B. helped in assembling data, manuscript writing and discussion. S.C., Y.K., A.U., S.R. and V.P. performed the experiments. S.G. provided intellectual inputs and reviewed the manuscript. N.T. recruited patients, collected biological samples and reviewed manuscript. D.B. conceived and supervised the study; critically evaluated and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Giri, A., Banerjee, P., Chakraborty, S. et al. Genome wide association study of uric acid in Indian population and interaction of identified variants with Type 2 diabetes. Sci Rep 6, 21440 (2016). https://doi.org/10.1038/srep21440
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21440
This article is cited by
-
Polygenic risk score trend and new variants on chromosome 1 are associated with male gout in genome-wide association study
Arthritis Research & Therapy (2022)
-
TRIM46 aggravated high glucose-induced hyper permeability and inflammatory response in human retinal capillary endothelial cells by promoting IκBα ubiquitination
Eye and Vision (2022)
-
Multifaceted genome-wide study identifies novel regulatory loci in SLC22A11 and ZNF45 for body mass index in Indians
Molecular Genetics and Genomics (2020)
-
Genome-wide association study of blood lipids in Indians confirms universality of established variants
Journal of Human Genetics (2019)
-
Genomewide association study of C-peptide surfaces key regulatory genes in Indians
Journal of Genetics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
