
Abstract
Inherited retinal disease (IRD) is a category of genetic disorders affecting retina. Understanding the molecular basis of IRD is vital for clinical and genetic classification of patients. Uyghur people is an isolated ethnic group mainly residing in northwestern China with genetic admixture from Europeans and East Asians. The genetic etiology of IRD in this specific population still remains unknown. Here, by next-generation sequencing (NGS), we screened mutations in over 200 known retinal disease genes in a cohort of 12 unrelated Uyghur IRD probands. Out of the 12 probands, six are solved with high confidence, two with low confidence, while the remaining four are unsolved. We identified known disease-causing alleles in this cohort that suggest ancient Uyghur migration and also discovered eight novel disease-associated variants. Our results showed NGS-based mutation screening as a reliable approach for molecular diagnosis. In addition, this approach can also be applied to reveal the genetic history of a specific ethnic group.
Similar content being viewed by others


Introduction
Retina is the structure surrounding the back of the vitreous body in the eye. It transforms light information into electrophysiological signals for human visual perceptions. The retina consists of various types of cells with highly specialized structures for its specific functions. Therefore, a number of pathways play important roles in the survival and functions of retina, including photo-transduction1, ciliogenesis2,3, metabolism4, transcription regulation5,6, vascular development7, protein folding8, etc. Correspondingly, mutations in genes involved in these pathways will result in a series of genetic disorders named inherited retinal disease (IRD), including retinitis pigmentosa (RP, MIM# 268000), Leber congenital amaurosis (LCA, MIM# 204000), cone-rod dystrophy (CRD, MIM# 120970), familial exudative vitreoretinopathy (FEVR, MIM# 133780), etc. IRD shows highly complex genetic etiology, with diverse inheritance patterns and more than two hundred disease-causing genes identified9.
With the advent of next-generation sequencing (NGS), numerous studies have applied NGS technology to reveal the molecular basis of human Mendelian disease including IRD. Specifically, customized target capture sequencing was used to screen mutations in known disease-causing genes with high efficiency7,10,11. By this method, novel disease-causing alleles as well as genotype-phenotype correlations have been identified, thus greatly enhancing our understanding of allele pathogenicity, protein function and population genetics7,10,11. Hence, NGS-based molecular diagnosis has been proven as a robust approach for assessing Mendelian disease in a molecular level.
Uyghur people are a Turkic language-speaking ethnic group mainly residing in Tarim Basin of northwestern China. Tarim Basin is an extremely isolated region due to the semi-arid or desert climate and its bordering rugged Karakoram, Kunlun and Tianshan mountain ranges. The Uyghur people were greatly influenced by Middle East Muslim culture after 10th century A.D. and they possess distinct genetic admixture derived from both East Asians and Europeans12. Previous genetic studies in Uyghur people mainly focused on complex diseases such as Type-2 diabetes, obesity, etc.13,14,15. However, NGS-based mutation screening in IRD cohorts is still missing. Thus, it is intriguing for us to understand the genetic etiology of IRD patients in this specific ethnic group.
Here, by NGS-based target capture sequencing, we performed molecular diagnosis in a cohort of 12 Uyghur IRD probands. This approach successfully solved six IRD cases with high confidence, achieving a 50% solving rate. We also identified disease-causing alleles that represent the migration history of Uyghurs and novel disease-associated variants, which may be ethnicity-specific.
Results
Twelve IRD probands were collected
A total of 12 well-characterized IRD probands of Uyghur ethnicity from Moyu County, Xinjiang, China were collected in this study. Of the 12 probands, nine are diagnosed with RP, two are LCA, and one is cone dystrophy. Eight out of twelve probands have multiple affected members in the same family, while the remaining four are simplex cases.
High quality capture sequencing data were generated
To identify disease-causing mutations in this IRD cohort, NGS-based target capture sequencing was performed on 12 probands. After the data acquisition and analysis, 95.2% of target regions had coverage of ≥10×, 88.3% of bases had coverage of ≥20×, and 63.4% of bases had coverage of ≥40×, indicating that sufficient sequencing coverage was achieved to enable high sensitivity for variant detection (Fig. 1). To make sure the sequencing coverage over the targeted regions was evenly distributed, evenness scores were calculated and the average is 0.8 for these 12 samples, suggesting a nearly uniform distribution of sequencing coverage.

Pathogenic mutations were identified in 6 probands with high confidence
An average of 663 raw variants, including 605 SNPs and 58 Indels were obtained initially (Table 1). After all filtering and annotation procedures, an average of 11.2 variants (10.5 SNPs and 0.7 Indels) per sample were identified (Table 1) and were assumed as candidate pathogenic variants. By analyzing these variants, we determined known and novel plausible pathogenic mutations in 8 probands. Out of them, six probands were solved with high confidence, conferring a solving rate of 50% (6/12).
RP cases
Uyg119 is the proband of an autosomal recessive RP (arRP) family with 3 affected members in the same generation (Figs 2A and 3A,B). She is 53 years old with a visual acuity of 0.01 (decimal scale) OU. NGS data showed that she possesses biallelic missense USH2A variants (Table 2). One variant (c.C842 > T, p.T281I) was reported as disease-causing in patients with Usher syndrome16. The other variant (c.G11815 > A, p.E3939K) is novel. This novel variant is extremely rare with a population frequency of 0.0006 and is predicted to be damaging by most algorithms, suggesting USH2A as the disease-causing gene in this family. We then performed Sanger sequencing to confirm the variants’ identity and co-segregation with the phenotype. The results showed that two unaffected members only have one variant, indicating these two variants are in trans. All affected individuals in this family don’t have hearing problems.

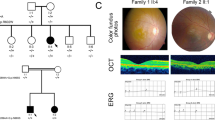
The genetic findings of the families Uyg119 (A), Uyg105 (B), Uyg132 (C), Uyg101 (D), Uyg131 (E), Uyg129 (F), Uyg125(G) and Uyg122 (H). mRNA accession ID of the disease-causing gene was labeled at the bottom of each pedigree. Genotypes (protein-altering changes based on the mRNA accession ID) were labeled next to the tested individuals. Arrows indicate the proband of each IRD family.

The Funduscopy images of probands Uyg119, OD (A) and OS (B); Uyg132, OD (C) and OS (D); Uyg122, OD (E) and OS (F); OCT results of the probands Uyg125, OD (G) and OS (H); Uyg122, OD (I) and OS (J).
Proband Uyg105 is a simplex RP case (Fig. 2B). She is 23 years old with a visual acuity of 0.12 OD and 0.08 OS. NGS data showed she has a homozygous stopgain variant (c.C4222 > T, p.Q1408*) in USH2A gene (Table 2). This variant is a known disease-causing allele for non-syndromic RP17. Sanger sequencing confirmed the homozygosity of this variant.
Uyg132 is the proband of an autosomal dominant RP (adRP) family with two affected individuals (Figs 2C and 3C,D). NGS identified a PRPF31 stopgain variant (c.C466 > T, p.Q156*) (Table 2). This variant is novel but its loss-of-function nature strongly suggests the pathogenicity. Sanger sequencing showed that the proband’s affected mother also possesses this variant and her unaffected sister does not, thus confirming the co-segregation.
Proband Uyg101 is in another adRP family with incomplete penetrance (Fig. 2D). NGS identified a heterozygous variant in the adRP gene SEMA4A (c.C1019G, p.A340G) (Table 2). The variant is absent in control databases. It affects a highly conserved amino acid site and considered to be damaging. Sanger sequencing confirmed this variant in the proband and another distantly-related affected family member, suggesting it as the putative disease-causing variant. Since only a limited number of adRP families with SEMA4A variants were reported before18,19, the causality and the pathogenicity of this variant need to be validated by further experiments evaluating its functional impact.
Proband Uyg131 is in a family ascertained with arRP or X-linked RP since there are two male affected individuals in the same generation (Fig. 2E). NGS data showed that the proband possesses an inframe deletion in RPGR (c.3225_3227delAGA, p.1076delE) (Table 2). This variant is absent in control databases, suggesting the rareness. The putative disease-causing gene RPGR also agrees with the potential X-linked inheritance pattern in this family. Sanger sequencing showed that both affected individuals are hemizygous for this allele. However, since it’s an inframe deletion, the pathogenicity of this variant is still unknown and needs to be confirmed by more genetic or functional data. Therefore, the disease association of this variant was considered of low confidence.
Proband Uyg129 is a simplex RP case (Fig. 2F). NGS identified a hemizygous missense variant in RPGR (c.G2572 > A, p.G858R) (Table 2). The variant is not found in any control databases. The predicted protein-altering effect is “low damaging” by MutationAssessor, while it is considered as “benign” or “unknown” by other algorithms. Thus, the pathogenicity of this variant needs further validation and we categorized its disease association as low confidence.
For the remaining three RP probands, Uyg108, Uyg115 and Uyg117, there are no plausible pathogenic variants identified by our target capture sequencing, suggesting the causative allele may reside in novel disease-causing genes or deep intronic regions.
LCA and cone dystrophy cases
Proband Uyg125 is a simplex LCA case (Figs 2G and 3G,H). NGS showed that the proband has a splicing variant (c.721-1G > A, p.?) and a deletion that creates a premature stop codon (c.1062_1068delCGAAAAC, p.Y354*) in LCA5 (Table 2). The splicing variant is not found in any databases and the deletion was reported before as disease-causing in an LCA family20. Sanger sequencing showed that the splicing variant was from the proband’s mother and the deletion was from the father. The proband’s unaffected brother only possesses the deletion variant.
Proband Uyg122 is in a recessive LCA family (Figs 2H and 3E,F,I,J). He is 41 years old with a visual acuity of 0.12 OD and 0.02 OS. NGS data identified compound heterozygous variants in TULP1, a known LCA-causing gene (Table 2). The first variant (c.C931 > T, p.R311W) is not found in any databases and is predicted to be pathogenic by all algorithms. The second variant (c.A1475 > G, p.Q492R) is also absent in controls and is considered damaging. Sanger sequencing showed both variants exist in two affected individuals and an unaffected sister only possesses one LCA5 variant, thus confirming the co-segregation.
Uyg111 is a 25-year-old male diagnosed with cone dystrophy. He has a visual acuity of 0.01 OU. Target capture sequencing identified no putative causative variants.
Discussion
In this study, we performed an NGS-based molecular diagnosis on 12 IRD probands of Uyghur ethnicity, including nine RP cases, two LCA cases and one cone dystrophy case. Our method solved 6 out of 12 probands with high confidence, achieving a solving rate of 50%. In addition, we also potentially solved two cases with low confidence.
Six probands, including four RP cases (Uyg119, Uyg105, Uyg132 and Uyg101) and two LCA cases (Uyg125, Uyg122) are considered to be solved with high confidence. Specifically, in the family of Uyg119, an USH2A variant previously reported to cause Usher syndrome17 was identified in our non-syndromic RP case. A plausible explanation for the genotype-phenotype correlation is that: the other USH2A variant (c.G11815 > A, p.E3939K) has a milder damaging effect, thus alleviating the overall impact on the phenotype. Consistent with this explanation, the novel USH2A variant is not considered damaging by all prediction algorithms, suggesting its hypomorphic nature instead of complete loss-of-function. Another explanation would be that the genetic background may modify the phenotype of patients.
Two RP probands (Uyg131 and Uyg129) are solved with low confidence. For these two cases, it is unknown if the RPGR variants confer pathogenicity given the unsureness of predicted damaging effect. Future functional data on RPGR mutant proteins are needed to clarify this question. Nevertheless, the rareness of these two variants supports the potential pathogenicity in a human genetics perspective.
Four of our probands do not have putative causative variants identified. The missing heritability could be from disease-causing mutations in a novel disease gene. Another possibility is that the disease-causing mutations lie in the non-coding regions of known disease genes and they cannot be detected by current NGS-based target capture sequencing. Future studies by whole exome sequencing or enhanced NGS approaches that target non-coding regions would help to understand the genetic basis of current unsolved cases.
All of the patients in this study are Uyghurs from Moyu County, a town located at the south rim of Tarim Basin where the residents are almost homogeneous of Uyghur origin. Uyghurs are the descendants of Turkic-speaking people who migrated to the Tarim Basin and several other ethnicities21. Contemporary studies suggest that they possess a distinct genetic admixture between Europeans and East Asians and have evolved for a long time12,22,23. In this study, we totally identified three known disease-causing variants (Table 2). Interestingly, one known USH2A mutation (c.C842T, p.T281I) was reported in a patient of Turkish origin16, supporting the ancient migration history. One LCA5 mutation we identified (c.1062_1068delCGAAAAC, p.Y354*), was previously reported in an Arabic Muslim family20, also suggesting the potential genetic influences from the Middle East. As for the third reported mutation (USH2A, c.C4222T, p.Q1408*), the patient ethnicity in previous literature was unknown17. We also identified several novel putative disease-causing variants, probably representing ethnicity-specific alleles. Further studies on larger cohorts might reveal more founder mutations that specifically exist in this ethnic group or Central Asia population. In addition, the cross-validation of the novel variants we identified in other cohorts could help us to infer the migration and evolution history of Uyghur ethnicity more accurately.
In summary, our approach identified the genetic cause of 50% IRD probands from an isolated ethnic group. A total of eight novel variants with potential pathogenicity were identified. Our study has revealed the molecular basis of IRD in Uyghurs and identified evidence of their genetic origin and migration history. Future NGS-based studies in more cohorts from this or adjacent regions will be of both scientific and cultural significance to improve our understanding of IRD molecular mechanism as well as genetic history of diverse ethnicities.
Methods
Clinical diagnosis of retinal disease patients
Uyghur IRD patients and other family members were ascertained at Minguang Ophthalmic Hospital (MOH), Hotan, Xinjiang, China. All patients provided written consent in accordance with the tenets of the Declaration of Helsinki. Comprehensive ophthalmic examinations including visual acuity testing (tumbling E chart), visual field testing (APS-6000, Xinchangzheng, Nanchang, China), optical coherence topography (OCT, 3D OCT-2000 Spectral Domain; Topcon, Tokyo, Japan) and funduscopy (APS-CER, Kanghua, Chongqing, China) were performed on each patient. Pedigrees were established by interviews. Genomic DNA was extracted from peripheral blood by using Qiagen kit (Qiagen Inc). All experimental methods were approved by the Institutional Review Boards of MOH and Chinese Academy of Sciences and were performed in accordance with relative guidelines and regulations.
Capture panel sequencing
We used customized target capture for NGS. The detailed information of the capture panel can be found in previous literature10. Briefly, 1 μg of patients’ genomic DNA was sheared into 300–500 bp fragments. After they were end-repaired, a single adenine base was added to 3′ ends. Then we applied ligation by using adapters before DNA fragments amplification. The DNA libraries were quantified using pico green assay kit (Invitrogen, Carlsbad, CA, USA). We then washed and recovered captured DNA by Agilent Hybridization and Wash Kit. Sequencing was performed on Illumina Hiseq 2000 (Illumina, San Diego, CA, USA).
Bioinformatics analysis
100 bp paired-end sequencing reads were obtained. Reads were aligned using BWA version 0.6.124. The Genome Analysis Tool Kit version 1.05974 was used for base quality recalibration and local realignment. Atlas-SNP2 and Atlas-Indel2 were used for calling SNPs and Indels. We obtained the variant frequencies from a series of public and internal control databases including Exome Aggregation Consortium (ExAC) database, CHARGE consortium25, ESP-650026 and 1000 Genome Project27. Since IRD are rare Mendelian disorders, variants with a frequency higher than 1/200 (for a recessive model) or 1/10,000 (for a dominant model) were filtered out. Then we excluded synonymous and deep intronic (distance >10 bp from exon-intron junctions) variants from further analysis. ANNOVAR (version 11/12/2014) and dbNSFP suite (version 2.9, contains SIFT, PolyPhen-2, LRT, MutationTaster, MutationAssessor, FATHMM, VEST3, etc.) were used to annotate and predict protein-altering changes. Known retinal disease-causing alleles were detected based on the HGMD professional database (version 11/15/2014).
Sanger validation and co-segregation test
Each putative disease-causing mutation was validated by Sanger sequencing. Primer3 was used to design a pair of primers to ensure that the amplicons cover 500 bp region around the mutation site. The PCR amplicons were Sanger sequenced on an ABI 3730XL Genetic Analyzer. The results were analyzed by Sequencher 5.0.
Additional Information
How to cite this article: Tajiguli, A. et al. Next-generation sequencing-based molecular diagnosis of 12 inherited retinal disease probands of Uyghur ethnicity. Sci. Rep. 6, 21384; doi: 10.1038/srep21384 (2016).
Change history
06 July 2016
The version of this Article previously published omitted Ruifang Sui and Rui Chen as corresponding authors. Correspondence and request for materials should also be addressed to hrfsui@163.com and ruichen@bcm.edu. This has now been corrected in the PDF and HTML versions of the Article.
References
Marlhens, F. et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat. Genet. 17, 139–141, doi: 10.1038/ng1097-139 (1997).
den Hollander, A. I. et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561, doi: 10.1086/507318 (2006).
Xu, M. et al. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum. Genet. doi: 10.1007/s00439-015-1586-x (2015).
Wang, F. et al. A missense mutation in HK1 leads to autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 55, 7159–7164, doi: 10.1167/iovs.14-15520 (2014).
Wang, F. et al. A Homozygous Missense Mutation in NEUROD1 Is Associated With Nonsyndromic Autosomal Recessive Retinitis Pigmentosa. Invest. Ophthalmol. Vis. Sci. 56, 150–155, doi: 10.1167/iovs.14-15382 (2015).
Freund, C. L. et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat. Genet. 18, 311–312, doi: 10.1038/ng0498-311 (1998).
Salvo, J. et al. Next-generation sequencing and novel variant determination in a cohort of 92 familial exudative vitreoretinopathy patients. Invest. Ophthalmol. Vis. Sci. 56, 1937–1946, doi: 10.1167/iovs.14-16065 (2015).
Xu, M. et al. ATF6 Is Mutated in Early Onset Photoreceptor Degeneration With Macular Involvement. Invest. Ophthalmol. Vis. Sci. 56, 3889–3895, doi: 10.1167/iovs.15-16778 (2015).
Daiger, S. P., Greenberg, J., Christoffels, A. & Hide, W. RetNet, Available at http://www.sph.uth.tmc.edu/RetNet/ (Accessed 15 Mar 2015.
Zhao, L. et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum. Genet. 134, 217–230, doi: 10.1007/s00439-014-1512-7 (2015).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 133, 331–345, doi: 10.1007/s00439-013-1381-5 (2014).
Xu, S., Huang, W., Qian, J. & Jin, L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am. J. Hum. Genet. 82, 883–894, doi: 10.1016/j.ajhg.2008.01.017 (2008).
Li, S. et al. Influences of APOA5 variants on plasma triglyceride levels in Uyghur population. PloS one 9, e110258, doi: 10.1371/journal.pone.0110258 (2014).
Song, M. et al. The Uyghur Population and Genetic Susceptibility to Type 2 Diabetes: Potential Role for Variants in CDKAL1, JAZF1, and IGF1 Genes. OMICS doi: 10.1089/omi.2014.0162 (2015).
Zhang, F., Xu, L., Jin, L. & Wang, X. F. A common variant in the FTO gene is associated with obesity in the Uyghur population. J. Endocrinol. Invest. 31, 1043 (2008).
Le Quesne Stabej, P. et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 49, 27–36, doi: 10.1136/jmedgenet-2011-100468 (2012).
Seyedahmadi, B. J., Rivolta, C., Keene, J. A., Berson, E. L. & Dryja, T. P. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp. Eye Res. 79, 167–173, doi: 10.1016/j.exer.2004.03.005 (2004).
Abid, A., Ismail, M., Mehdi, S. Q. & Khaliq, S. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J. Med. Genet. 43, 378–381, doi: 10.1136/jmg.2005.035055 (2006).
Glockle, N. et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 22, 99–104, doi: 10.1038/ejhg.2013.72 (2014).
Beryozkin, A. et al. Identification of mutations causing inherited retinal degenerations in the israeli and palestinian populations using homozygosity mapping. Invest. Ophthalmol. Vis. Sci. 55, 1149–1160, doi: 10.1167/iovs.13-13625 (2014).
Lattimore, O. Return to China’s Northern Frontier. Geogr. J. 139, 233–242, doi: 10.2307/1796091 (1973).
Li, H., Cho, K., Kidd, J. R. & Kidd, K. K. Genetic landscape of Eurasia and “admixture” in Uyghurs. Am. J. Hum. Genet. 85, 934–937; author reply 937–939, doi: 10.1016/j.ajhg.2009.10.024 (2009).
Yao, Y. G., Kong, Q. P., Wang, C. Y., Zhu, C. L. & Zhang, Y. P. Different matrilineal contributions to genetic structure of ethnic groups in the silk road region in china. Mol. Biol. Evol. 21, 2265–2280, doi: 10.1093/molbev/msh238 (2004).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi: 10.1093/bioinformatics/btp324 (2009).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2, 73–80, doi: 10.1161/circgenetics.108.829747 (2009).
Tennessen, J. A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69, doi: 10.1126/science.1219240 (2012).
Abecasis, G. R. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073, doi: 10.1038/nature09534 (2010).
Acknowledgements
We thank all the patients and their family members for participating in this study. We thank the Exome Aggregation Consortium and the groups that provided exome variant data. NGS was conducted at the Functional Genomic Core (FGC) facility at Baylor College of Medicine supported by NIH shared instrument grant 1S10RR026550 to R.C. This study was funded by the Projects of the Central Asian Drug Discovery & Development Centre of Chinese Academy of Sciences (Grant No. 201406). This work was also supported by grants from National Eye Institute (R01EY022356, R01EY018571), Retinal Research Foundation, Foundation Fighting Blindness (BR-GE-0613-0618-BCM) to R.C. M.X. is supported by Cullen Foundation endowment to the Molecular and Human Genetics Graduate Program, Baylor College of Medicine.
Author information
Authors and Affiliations
Contributions
A.T., M.X., R.C. and H.A.A. designed the study and experiments. A.T., Q.F., R.S., R.Y., A.E. and H.A.A. collected clinical data. K.W. and Y.L. performed NGS and Sanger sequencing experiments. A.T. and M.X. analyzed the sequencing data. A.T. and M.X. wrote the main manuscript text and prepared all the figures. All authors reviewed and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tajiguli, A., Xu, M., Fu, Q. et al. Next-generation sequencing-based molecular diagnosis of 12 inherited retinal disease probands of Uyghur ethnicity. Sci Rep 6, 21384 (2016). https://doi.org/10.1038/srep21384
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21384
This article is cited by
-
Whole-exome sequencing in 168 Korean patients with inherited retinal degeneration
BMC Medical Genomics (2021)
-
Clinical exome sequencing facilitates the understanding of genetic heterogeneity in Leber congenital amaurosis patients with variable phenotype in southern India
Eye and Vision (2021)
-
Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
