
Abstract
The activation/inactivation of HIF1α is precisely regulated in an oxygen-dependent manner. HIF1α is essential for hypoxia induced apoptosis and cell cycle arrest. Several recent studies indicated that the expression of miRNAs can be modulated by hypoxia. However, the involvement of miRNAs in the regulation of HIF1α induction remains elusive. In present study, we demonstrated that miR-101 was rapidly and transiently induced after hypoxia in breast cancer cells. Over-expression of miR-101 significantly inhibited cell proliferation in breast cancer cells through increased apoptosis and cell cycle arrest in normoxia condition. This inhibitory phenomenon seems due to miR-101-mediated induction of HIF1α, because we identified that VHL, a negative regulator of HIF1α, is a novel target of miR-101 and over-expression of miR-101 decreased VHL levels and subsequently stabilized HIF1α and induced its downstream target VEGFA. Furthermore, we demonstrated that siRNA-mediated knockdown of VHL or HIF1α overexpression could also induce apoptosis and cell cycle arrest whereas enforced expression of VHL, administration of anti-miR-101 oligos or treatment of 2-MeOE2, an inhibitor of HIF1α, could rescue cells from such inhibition. These results reveal a novel regulatory mechanism of HIF1α induction in normoxia and suggest that miR-101 mediated proliferation inhibition may through HIF1α mediated apoptosis and cell cycle arrest.
Similar content being viewed by others

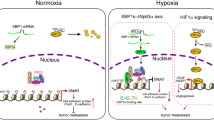

Introduction
Hypoxia is one of the classical features in solid tumors. Hypoxia inducible factor-1 (HIF1), a heterodimer with HIF1α and HIF1β subunits, is the major activated transcriptional factor in response to hypoxia. Although HIF1β is constitutively expressed, the protein level of HIF1α is tightly regulated by the oxygen levels. When oxygen is adequate, HIF1α is hydroxylated by prolyl hydroxylase proteins (PHDs) and in turn recognized by the von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase(VHL) for unbiquitin-mediated degradation1. Under hypoxia, the hydroxylase activity of PHDs is dramatically reduced; thus HIF1α is stabilized, dimerized with HIF1β to form HIF1 and translocated to the nucleus2. Because the increased protein level of HIF1α is frequently observed and correlated with poor prognosis in many cancer types, a long-lasting concept believes that HIF1α plays an oncogenic role in tumor growth3,4. However, some recent well-designed studies challenged this concept. Carmeliet et al. showed that HIF1α is required for hypoxia-mediated apoptosis and inhibition of cell proliferation5. HIF1α deficiency apparently inhibited tumor growth and enhanced tumor invasion in microenvironment with sufficient oxygen supply6. HIF1α stabilization due to loss of VHL decreased tumor growth7. Furthermore, HIF1α interacted with Myc or Cdc6 to induce cell cycle arrest in the absence of hypoxic signal8. Therefore, the role of HIF1α in tumor progression has been controversial.
MicroRNAs are evolutionally conserved short noncoding RNAs that negatively regulate the expression of both protein-coding and noncoding genes9,10. Due to the partial complement to their targets, a microRNA can regulate multiple genes’ expression simultaneously11. Recent studies have indicated that hypoxia modulates the expression of a specific set of microRNAs, termed “hypoxamirs”12,13,14,15. Given adaptation to hypoxia is essential for solid tumor progression, it’s intriguing to explore the feedback regulatory loops between “hypoxamirs” and hypoxic pathway. In this regard, several hypoxamirs have been shown to regulate hypoxic pathway in either negative or positive feedback loops. For example, by directly targeting HIF1α, two hypoxamirs, miR-20b and miR-199a, suppress hypoxia progression16,17. miR-424, induced by hypoxia, targets cullin 2 (CUL2) to stabilize HIF1α and enhance angiogenesis18.
miR-101 is evolutionally conserved in vertebrates. Down-regulation of miR-101 has been observed in various types of cancers, including prostate, colon, bladder, gastric, ovarian and breast cancers19,20,21,22,23,24,25. Ectopic expression of miR-101 has been shown to induce cell apoptosis and inhibit cell proliferation26,27. However, our previous study demonstrated that enforced expression of miR-101 confers cells estrogen-independent growth ability in breast cancer28. A recent report also showed that increased miR-101 levels are negatively associated with the overall survival (OS) and disease-free interval (DFI) in patients with ovarian carcinoma and miR-101 could target C-terminal binding protein-2 (CtBP2) to enhance the stemness of cancer cells29. These controversial results indicate that miR-101 has divergent and even antagonistic roles in tumor progression. Interestingly, Kim JH et al. showed that hypoxia could induce miR-101 expression in human umbilical vein endothelial cells (HUVECs), human brain microvascular endothelial cells (HBMECs), astrocytes, HeLa and U937 cells30, suggesting that miR-101 is a novel hypoxamir under the regulation of hypoxia. However, little is known about the roles of miR-101 in regulating the components of hypoxia pathway.
In this study, we demonstrated that miR-101 is an acute miRNA in response to hypoxia in breast cancer cells. miR-101 could directly target VHL, a negative regulator of HIF1α, resulting in HIF1α induction in the absence of hypoxic signal. miR-101 directed proliferation inhibition seems due to HIF1α mediated apoptosis and cell cycle arrest.
Results
Hypoxia induced miR-101 expression in breast cancer cells
Since Kim JH et al. showed induction of miR-101 in Hela and U937 cells 12 h after hypoxia, we then examined whether hypoxia could induce miR-101 expression in breast cancer cells. MCF-7 and MDA-MB-231 cells were exposed to hypoxia condition (1% Oxygen) and miR-101 levels were measured by TaqMan qPCR at indicated time points. Figure 1A showed that miR-101 expression rapidly reached a peak at 1 h after hypoxia exposure and then declined to the basal level in 24 h in MCF-7 cells (Fig. 1A). We also observed similar kinetic changes but with much less induction of miR-101 in MDA-MB-231 cells after hypoxia (data not shown). Consistent with previous reports, the expression of VEGFA, a well known downstream target of hypoxia, was dramatically induced after hypoxia (Fig. 1B). Western blot showed that the induction of HIF1α started in 1 h, reached its peak at 6 h and declined in 24 h after hypoxia (Fig. 1C). The induction of VEGFA was also confirmed by western blot (Fig. 1C). To further investigate the correlation between hypoxia and miR-101, we treated cells with the hypoxia-mimetic desferrioxamine (DFO). As shown in Fig. 1D,E, DFO treatment significantly increased HIF1α and VEGFA levels in both MCF-7 and MDA-MB-231 cells. Interestingly, unlike the rapid induction of miR-101 in hypoxia treatment, miR-101 levels increased at 2 h and 6 h after DFO exposure in MCF-7 and MDA-MB-231 cells respectively (Fig. 1D,E).

miR-101 is induced upon hypoxia exposure.
(A) miR-101 levels in MCF-7 after hypoxia exposure by qPCR. (B) VEGFA induction in MCF-7 and MDA-MB-231 cells by qPCR. (C) HIF1α and VEGFA induction after hypoxia by western blot. (D,E) HIF1α and VEGFA induction by western blot and miR-101 expression by qPCR in MCF-7 (D) or MDA-MB-231 cells (E) after DFO treatment. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
miR-101 inhibits the cell proliferation in breast cancer cells
To examine the functions of this novel hypoxamir in breast cancer cells, we stably expressed miR-101 in MCF-7 and MDA-MB-231 cells after lentiviral infection. The ectopic expression of miR-101 was confirmed by TaqMan qPCR in both cell lines (data not shown). Cell proliferation assay was performed. Figure 2A,B showed that over-expression of miR-101 significantly inhibited the cell proliferation in both cell lines. To examine whether miR-101 inhibits clonogenic survival, 2-dimension colony formation assay was performed. Compared to the control group, over-expression of miR-101 significantly suppressed the colony formation to about 35% and 26% in MCF-7 and MDA-MB-231 cells respectively (Fig. 2C). To further assess whether miR-101 suppresses cell proliferation, Edu incorporation was examined. Compared to the control group, miR-101 expression dramatically impaired the incorporation of Edu in both MCF-7 and MDA-MB-231 cells (Fig. 2D).

miR-101 inhibits cell proliferation.
(A,B) Cell proliferation in MCF-7 (A) and MDA-MB-231 cells (B) with or without miR-101 over-expression. (p < 0.0001 by two-way ANOVA). (C) Clonogenic survival in both MCF-7 and MDA-MBA-231 cells with or without miR-101 over-expression. (D) Edu incorporation in MCF-7 and MDA-MB-231 cells with or without miR-101 over-expression. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
miR-101 induces apoptosis and cell cycle arrest in breast cancer cells in normoxia
Since apoptosis and cell cycle arrest are tightly associated with reduced cell proliferation, we then determine whether miR-101 could affect the apoptosis and cell cycle in breast cancer cells. Annexin V and Propidium iodide (PI) staining indicated that miR-101 significantly induced cell apoptosis in both MCF-7 and MDA-MB-231 cells (Fig. 3A). DAPI staining further confirmed the miR-101-induced apoptosis in both cell lines (Fig. 3B). PI/FACS analysis demonstrated that miR-101 over-expression caused significant G1-phase arrest in both cell lines (Fig. 3C). In addition, we examined the effects of miR-101 on apoptosis and cell cycle of MCF-7 cells in hypoxia condition, but no significant changes were observed (Fig. S1). Given the essential role of CyclinD1 in regulating cell cycle, we started to check if miR-101 mediated cell cycle arrest is due to the changes of CyclinD1. qPCR and western blot showed that neither mRNA nor protein levels of CyclinD1 was affected by miR-101 in either normoxia or hypoxia condition (Fig. S2).

miR-101 induces apoptosis and cell cycle arrest.
(A) Apoptosis assay in both MCF-7 and MDA-MB-231 cells with or without miR-101 over-expression by Annexin V and PI double staining followed with flow cytometry analysis. (B) DAPI staining to detect apoptotic cells. (C) Cell cycle analysis in both MCF-7 and MDA-MB-231 cells with or without miR-101 over-expression by PI staining followed by flow cytometry analysis. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
VHL is a direct target of miR-101
Previous studies have demonstrated that miR-101 targets multiple genes, such as EZH2, COX-2, STMN1, ROCK2 and so on, to inhibit tumor growth19,20,31,32. In this study, we focused on exploring the putative regulation loops between miR-101 and hypoxia pathway. After miRNA database search, we identified that VHL, a negative regulator of HIF1α, is a putative target of miR-101 (miRDB v4.0). As shown in Fig. 4A, the VHL-3′UTR carries two putative binding sites of miR-101. We constructed a luciferase reporter vector carrying a VHL 3′UTR region containing those two binding sites. Compared to vector control, over-expression of miR-101 significantly inhibited luciferase activity of VHL-3′UTR down to 40% as well as had no effect on the luciferase activity of pGL3-control vector (Fig. 4B). Figure 4C showed that miR-101 mediated reduction of luciferase activity was in a dose-dependent manner. To assess the specificity of miR-101 mediated suppression, we employed anti-miR-101 oligos to block the suppression of miR-101. Compared to the negative control oligo, introduction of the anti-miR-101 reversed the miR-101-mediated suppression of luciferase activity (Fig. 4D). To further confirm whether miR-101 mediated suppression of luciferase activity is attributed to the direct binding of miR-101 to the VHL-3′UTR, we mutated those two putative binding sites individually or both. miR-101 induced suppression was significantly attenuated in VHL-3′UTR vectors with either site 1 or site 2 mutation and was completely abolished in VHL-3′UTR with site1 and site2 double mutations (Fig. 4E). These results indicated that VHL is a direct target of miR-101. To examine if miR-101 also targets on other negative regulators of hypoxia pathway, such as PHD1, PHD2, PHD3 and PHD4, we did an extensive search for putative miR-101 targets by employing a prediction program with less stringency (FINDTAR3: http://bio.sz.tsinghua.edu.cn/). FINDTAR3 indicated putative bindings between miR-101 and all those four negative regulators (Fig. S3). However, luciferase reporter assay failed to validate those interactions (Fig. S3). To determine whether miR-101 could directly downregulate endogenous VHL expression, we performed western blot in MCF-7 and MDA-MB-231 cells with stable ectopic expression of miR-101. As shown in Fig. 4F, miR-101 significantly suppressed VHL protein levels in both cell lines and this suppression was through post-transcriptional regulation because miR-101 over-expression did not reduce VHL mRNA levels (Fig. 4G). In response to VHL downregulation, the expression of HIF1α and VEGFA was increased (Fig. 4F).

VHL is a direct target of miR-101.
(A) Two putative binding sites of miR-101 on the 3′UTR of VHL. (B) Suppression of the luciferase activity by miR-101. (C) Suppression of the luciferase activity by miR-101 in a dose dependent manner. (D) anti-miR-101 increased the luciferase activity in cells with miR-101 over-expression. (E) Effect of miR-101 on the luciferase activity of VHL-3′UTR, VHL-3′UTR-m1, VHL-3′UTR-m2 and VHL-3′UTR-dm. (F) HIF1α, VEGFA, VHL level changes in MCF-7 and MDA-MB-231 cells with or without miR-101 over-expression by western blot. (G) mRNA level changes of VHL in MCF-7 and MDA-MB-231 cells with or without miR-101 over-expression by qPCR. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Down-regulation of VHL induces increased apoptosis and cell cycle arrest
To answer whether miR-101-mediated apoptosis and cell cycle arrest are due to VHL suppression, we employed VHL siRNA to specifically knockdown VHL expression. Realtime PCR and western blot validated successful knockdown of VHL in both mRNA and protein levels respectively in MCF-7 and MDA-MB-231 cells 48 hours after siVHL transfection (Fig. 5A,B). The consequent increase of HIF1α and VEGFA was also detected in cells with VHL suppression (Fig. 5B). Apoptosis and cell cycle assays were performed 48 hours post siVHL transfection. As shown in Fig. 5C–E, downregulation of VHL induced significant apoptosis and G1-phase arrest in MCF-7 and MDA-MB-231 cells compared to the negative control oligo.

Knockdown of VHL induces apoptosis and cell cycle arrest.
(A) VHL levels in MCF-7 and MDA-MB-231 cells transfected with siVHL by qPCR. (B) Protein level changes of VHL, HIF1α and VEGFA in MCF-7 and MDA-MB-231 cells transfected with siVHL by western blot. (C–E) Percentage of Annexin V positive cells (C), G1, S and G2/M phages (D) and G1 phase (E) in MCF-7 and MDA-MB-231 cells transfected with siVHL. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Overexpression of VHL rescues cells from miR-101-mediated apoptosis and cell cycle arrest
To further test whether miR-101 functions upstream of VHL to induce cell apoptosis and G1-phase arrest, we over-expressed VHL in both cell lines with stable ectopic miR-101 expression (Fig. 6A). Annexin V and PI staining showed that VHL over-expression significantly blocked miR-101 induced apoptosis in both cell lines (Fig. 6B). FACS assay indicated that miR-101 induced G1-phase arrest was significantly released upon VHL over-expression (Fig. 6C,D).

VHL restoration rescues cells from miR-101-mediated apoptosis and cell cycle arrest.
(A) Ectopic expression of VHL in MCF-7-miR-101 and MDA-MB-231-miR-101 cells by western blot. (B) Apoptosis assay in both MCF-7-miR-101 and MDA-MB-231-miR-101 cells with or without VHL over-expression by Annexin V and PI double staining followed with flow cytometry analysis. (C,D) Percentage of G1, S and G2/M phages (C) and G1 phase (D) in MCF-7-miR-101 and MDA-MB-231-miR-101 cells with or without VHL over-expression. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Anti-miR-101 oligos block miR-101 mediated apoptosis and cell cycle arrest
To answer whether inhibition of miR-101 could rescue cells from miR-101 induced apoptosis and cell cycle arrest, we transiently administrated anti-miR-101 oligos into cells with stable miR-101 over-expression. Compared with the negative control, released VHL repression was observed in cells transfected with anti-miR-101 (Fig. 7A). Cell aopotosis and FACS assays showed that blocking of miR-101 significantly rescued cells from miR-101 induced apoptosis and G1 phase arrest in both breast cancer cell lines (Fig. 7B–D).

Blocking of miR-101 releases cells from apoptosis and cell cycle arrest.
(A) VHL level changes in MCF-7-miR-101 and MDA-MB-231-miR-101 cells transfected with anti-miR-101 oligos by western blot. (B) Percentage of Annexin V positive cells in MCF-7-miR-101 and MDA-MB-231-miR-101 cells transfected with anti-miR-101 oligos. (C,D) Percentage of G1, S and G2/M phages (C) and G1 phase (D) in MCF-7-miR-101 and MDA-MB-231-miR-101 cells. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
HIF1a is required for miR-101 mediated apoptosis and cell cycle arrest
To examine whether HIF1a over-expression could induce apoptosis, MCF-7 cells with stable HIF1a over-expression were generated after lentiviral infection. Ectopic HIF1a expression was confirmed by western blot (Fig. 8A). Annexin V and PI staining indicated that HIF1a over-expression increased the percentage of apoptosis cells to near 2-fold (Fig. 8C). To determine if HIF1a is required for miR-101 induced apoptosis and cell cycle arrest, we employed 2-MeOE, an inhibitor of HIF1a, to treat MCF-7 cells with stable miR-101 over-expression. 2-MeOE2 successfully inhibited HIF1a activity as the VEGFA level was significantly decreased upon 500nM 2-MeOE2 treatment for 48h (Fig. 8B). As shown in Fig. 8D–F, inhibition of HIF1α significantly rescued cells from miR-101 mediated apoptosis and G1 phase arrest. Interestingly, consistent with a previous report that loss of HIF1α promotes G1 to S phase transition, MCF-7-miR-101 also showed significantly increased G1 to S progression upon 2-MeOE2 treatment (Fig. 8G).

HIF1α is attributed to miR-101 mediated apoptosis and cell cycle arrest.
(A) HIF1a level change in MCF-7 cells infected with HIF1a expression vector by western blot. (B) VEGFA level change in MCF-7-miR-101 cells after 48 h 2-MeOE2 treatment by qPCR. (C) Apoptosis assay in MCF-7 cells with HIF1a over-expression by Annexin V and PI double staining followed with flow cytometry analysis. (D) Apoptosis assay in MCF-7-miR-101 cells after treated with 2-MeOE2. (E–G) Percentage of G1, S and G2/M phages (E), G1 phase (F) and S phase (G) in MCF-7-miR-101 cells after 2-MeOE2 treatment. Data were analyzed using Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In present study, we demonstrated that miR-101 is a novel hypoxamir in breast cancer cells, whose expression was rapidly and transciently induced in 1 hour after hypoxia. Through directing cell apoptosis and cell cycle arrest, ectopic expression of miR-101 suppressed proliferation in breast cancer cells. To our knowledge, we for the first time identified that VHL is a direct target of miR-101 and demonstrated that miR-101 could increase HIF1α protein levels by repressing VHL in normoxia condition. Furthermore, we showed that an increase of HIF1a levels by either direct HIF1a over-expression or by indirect knockdown of VHL, a negative regulator of HIF1a, can induce apoptosis and cell cycle arrest. In contrast, HIF1a downregulation by VHL over-expression, anti-miR-101 oligo administration or 2-MeOE2 treatment rescued cells from miR-101-mediated apoptosis and cell cycle arrest in breast cancer cells.
Hypoxic microenvironment commonly exists in many solid tumors and is tightly associated with poor prognosis and resistance to therapy. HIF1α expression is induced in cells under hypoxia exposure and maintained at a basal level under normoxia4. Either the normoxic silencing or the hypoxic induction of HIF1α is orchestratedly modulated by multiple negative or positive regulators33. Hypoxia induced HIF1α dimerizes with HIF1β to transcript the expression of hundreds of downstream genes. miRNAs, a class of novel negative regulators of gene expression, have emerged in the downstream targets of HIF1α under hypoxia. These hypoxia mediated miRNAs are termed hypoxamiRs, including miR-210, miR-10b, miR-155, miR-372/373 and miR-424 whose expression was up-regulated and miR-199a, miR-20b and miR-200b whose expression was down-regulated under hypoxia16,17,18,34,35,36,37,38,39,40,41. Interestingly, some of those hypoxamiRs, such as miR-20b, miR-199a and miR-424, can form negative or positive feedback loops to modulate hypoxia pathway16,17,18.
miR-101, a conserved miRNA, was first reported as a tumor suppressor in prostate cancer. Thereafter, accumulating evidence had demonstrated the tumor suppression feature of miR-101 in many other tumor types including colon cancer, hepatocellular carcinoma, bladder cancer, gastric cancer, non-small cell lung cancer, breast cancer, pancreatic cancer and esophageal cancer20,21,22,25,26,42,43,44. However, by adopting an unbiased miRNA library screening system, we previously demonstrated that over-expression of miR-101 conferred cells estrogen-independent growth ability in MCF-7 and T47D cells, suggesting the oncogenic potential of miR-101 in breast cancer28. Interestingly, the oncogenic role of miR-101 has been recently reported by Dr. Weiping Zou’s group in ovarian cancer29. They demonstrated that Myeloid-derived suppressor cells (MDSCs) in ovarian carcinoma could induce the expression of miR-101 and in turn enhance the stemness of cancer cells29. Thus, these controversial data indicate that miR-101 plays a dual role as either a tumor suppressor or an oncogene in tumor progression. In present study, we demonstrated that miR-101 was induced at the very early stage of hypoxia and miR-101 could target VHL, resulting in stabilization of HIF1α in normoxia condition.
Emerging evidence has indicated that HIF1a has differential roles in tumor progression greatly dependent on the microenvironment of the tumor6. Blouw found that HIF1α deficient astrocytomas showed greater proliferation and invasion abilities when inoculated into the brain their natural habitat (with sufficient oxygen supplement) instead of the subcutaneous habitat (a poorly vascularized environment)6, suggesting the tumor suppressive role of HIF1a in the tumor with adequate oxygen supply. In gliomas with mutations in isocitrate dehydrogenase 1/2 (IDH1)IIDH2, HIF1α also presents as a tumor suppressor45. Koivunen found that the most common IDH mutant IDHR132H can activate PDHs to suppress HIF1α level, in turn promoting the proliferation and anchorage- independent growth abilities of immortalized astrocytes45. Therefore, HIF1α plays a suppressive role when the microenvironment of tumor is oxygen sufficient. Consistent with these previous findings, we demonstrated that miR-101 can target VHL, an inhibitor of HIF1α, to increase HIF1α level and in turn suppress cell proliferation via HIF1α mediated apoptosis and cell cycle arrest. Our finding suggested that the miR-101/VHL/HIF1α axis may play a tumor suppressive role when adequate oxygen was supplied to the tumor.
A recent paper reported that HIF1α plays a positive role in the proliferation in MDA-MB-231 cells46, which is opposite to our findings. However, it’s not convincing that Shi just employed a simple approach, siRNA mediated HIF1a knockdown, in their study to address the positive role of HIF1α. It’s also inappropriate for Shi to use only one siRNA duplex against HIF1a, because the phenotypes in Shi’s study may be probably caused by off-targeting.
In fact, several previous studies had also indicated the connection between miR-101 and hypoxia pathway. For example, miR-101 induction has been observed in HUVECs, HBMECs, astrocytes, HeLa and U937 cells under hypoxia treatment30; inhibition of miR-101 could rescue cardiomyocytes from apoptosis induced by hypoxia/reoxygenation47. Thus, previous data and the present study strongly implicated that miR-101 mediated inhibition on cell proliferation in breast cancer cells seems due to HIF1α-induced apoptosis and cell cycle arrest. Recently, miR-101 based targeted therapy had been reported in treatment of hepatocellular carcinoma in a mouse model with a great success32. However, when we appreciate the great potential of miR-101 in tumor therapy, necessary cautions must be emphasized given the dual role of miR-101 in tumor progression.
Methods
Cell Culture
HEK293T and human breast cancer cell lines MDA-MB-231 and MCF-7 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were maintained in DMEM medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 0.1 mg/ml Penicillin-Streptomycin (Gibco) and incubated at 37 °C in a humidified chamber supplemented with 5% CO2.
Hypoxia exposure
Cells were maintained in a hypoxic chamber flushed with a gas mixture containing 94%N2 and 5%CO2. Under these conditions, O2 levels in the medium were determined to be 1%. For DFO treatment, cells were treated with DFO (500 uM). Protein and RNA were isolated at indicated time points.
Plasmid construction and transfection
HIFα over-expression vector was purchased from Addgene (Plasmid# 19365). To generate miR-101 expression vectors, a 420 bp fragment carrying pre-miR-101 was amplified from the MCF-7 genomic DNA by the high fidelity polymerase Phusion enzyme (New England Biolabs, Ipswich, MA) using PCR primers:
5′ tctagaTATTTCAGCCTCACCACTTGCT
5′ tctagaCCCCATGTTACAAAACAAGGCA.
The amplified fragment was cloned into the pLVX-IRES-ZsGreen vector (Clontech) at the Xba1 site.
To generate the luciferase reporter vector carrying the VHL-3′UTR region carrying the two putative binding sites of miR-101, we amplified a 2243 bp VHL-3′UTR region from the genome DNA of MCF-7 by the high fidelity polymerase Phusion enzyme (New England Biolabs, Ipswich, MA) using PCR primers:
5′ tctagaGGAGTAGCCTGGACTGTTTCAT
5′ tctagaTCCTTGGACAACACCAAAAACAC
The amplified fragment was cloned into the pGL3-control vector (Promega) at the Xba1 site.
To generate the VHL-3′UTR reporter vectors with mutated miR-101 binding sites, we used Phusion Site-Directed Mutagenesis Kit (Life Technologies) to directly mutate those two binding sites by using primers:
VHL-UTR-mut1f: p-catagttgagattCACACACtcatacagtttta
VHL-UTR-mut1r: p-taaaaaccaaccaaaatctgccctaaa
VHL-UTR-mut2f: p-acatgccgtttgaCACACACgtttttggtgttg
VHL-UTR-mut2r: p-tttttttttgtttttttggtttctttttg
VHL siRNA and anti-miR-101 oligos were purchased from Shanghai GenePharma.
Quantitative real time-polymerase chain reactions (qRT-PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen). RNA was reverse-transcribed by RevertAidFirst Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR Green PCR real-time PCR Master MIX (Toyobo). miRNA was isolated using mirVana™ miRNA Isolation Kit(Life Technologies). The TaqMan® microRNA reverse transcription kit (Applied Biosystems) was used to synthesize the cDNA for miR-101. The expression level of U6 snRNA was used as an internal control for normalization.
Cell proliferation assay
20000/well cells were seeded into a six-well plate and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS), changing half of the medium every other day. Then counting cell number at indicated time points.
Luciferase assay
The dual-luciferase assay was performed 48 h after transfection according to the manufacturer’s instructions (Promega, Madison, WI, USA). The Renilla luciferase was used as a control reporter. The ratio of Renilla luciferase to firefly luciferase was normalized to the negative control. Three independent experiments were performed.
Apoptosis assay
Cells were harvested and washed twice with pre-cooled PBS and then were incubated in the binding buffer with a mixture containing annexin V (Biolegend) and propidium iodide (Sungene Biotech ) for 15 min in darkness. Cells were measured by flow cytometry. Three independent experiments were performed.
Cell cycle assay
The cells were fixed in chilled 75% ethanol and stained with a propidium iodine (PI) solution contains 100 ug/ml RNase (Tiangen Biotech) and 50 ug/ml PI (Biolegend) in PBS for cell cycle analysis.
Colony formation assay
1000/well cells were seeded into a six-well plate and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) for 14 days, changing half of the medium every other day. Colonies were fixed in 4% paraformaldehyde for 20 min at room temperature and then stained with crystal violet for 30 min at room temperature. The numbers of colonies were counted and pictures were captured.
Western blot
Cells were collected and lysed in Radio-Immunoprecipitation Assay (RIPA) buffer (Thermo Fisher, Rockford, IL, USA) containing 0.02% complete Protease Inhibitor EASY packs EDTA-Free (Roche Applied Science). Then the protein was separated using 12% polyacrylamide gel and transferred to polyvinyl difluoride membranes. After blocking with 5% bovine serum albumin (BSA) in TBS/Tween20 (TBST) and incubated with specific primary antibodies at 4 °C overnight, followed by a 5 min wash with TBS-T, which was repeated three times. After this, the membranes were incubated with horseradish peroxidase conjugated anti-mouse or anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at room temperature for 1 h, followed a 5 min wash with TBST, which was repeated three times. Then the membranes were analyzed with the ECL chemiluminescent detection system (Bio-Rad).
The following antibodies were used in this study: anti-actin monoclonal (Santa Cruz Biotechnology, Santa Cruz, CA, USA) , anti-HIF1α polyclonal (Cell Signaling Technology), anti-VEGFA polyclonal, anti-cyclinD1 polyclonal (ProteinTech Group, Chicago, IL, USA) and anti-VHL polyclonal (Cell Signaling Technology).
EdU (5-ethynyl-2’-deoxyuridine) incorporation assay
Cells were seeded on cover slips for 48 h. The assay was performed with EdU cell proliferation assay kit (Thermo Fisher, Rockford, IL, USA) according to the manufacturer’s instructions. Briefly, EdU was added to the culture media for 2 h and the final concentration was 10 μM. After labeling, cells were fixed in 4% formaldehyde. Then stained cells with Edu and counterstained with Hoechest, mounted in standard mounting media and imaged by fluorescence microscopy.
Additional Information
How to cite this article: Liu, N. et al. MicroRNA-101 targets von Hippel-Lindau tumor suppressor (VHL) to induce HIF1α mediated apoptosis and cell cycle arrest in normoxia condition. Sci. Rep. 6, 20489; doi: 10.1038/srep20489 (2016).
References
Forristal, C. E., Wright, K. L., Hanley, N. A., Oreffo, R. O. & Houghton, F. D. Hypoxia inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. Reproduction 139, 85–97 (2010).
Kaelin, W. G., Jr. & Ratcliffe, P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30, 393–402 (2008).
Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012).
Young, R. M. & Simon, M. C. Untuning the tumor metabolic machine: HIF-alpha: pro- and antitumorigenic? Nat Med 18, 1024–1025 (2012).
Carmeliet, P. et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394, 485–490 (1998).
Blouw, B. et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 4, 133–146 (2003).
Mack, F. A. et al. Loss of pVHL is sufficient to cause HIF dysregulation in primary cells but does not promote tumor growth. Cancer cell 3, 75–88 (2003).
Koshiji, M. et al. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J 23, 1949–1956 (2004).
Ambros, V. The functions of animal microRNAs. Nature 431, 350–355 (2004).
Leucci, E. et al. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci Rep 3, 2535 (2013).
Brennecke, J., Stark, A., Russell, R. B. & Cohen, S. M. Principles of microRNA-target recognition. PLoS Biol 3, e85 (2005).
Chan, S. Y. & Loscalzo, J. MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle 9, 1072–1083 (2010).
Kulshreshtha, R. et al. A microRNA signature of hypoxia. Mol Cell Biol 27, 1859–1867 (2007).
Giannakakis, A. et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol Ther 7, 255–264 (2008).
Hua, Z. et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PloS One 1, e116 (2006).
Rane, S. et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res 104, 879–886 (2009).
Cascio, S. et al. miR-20b modulates VEGF expression by targeting HIF-1 alpha and STAT3 in MCF-7 breast cancer cells. J Cell Physiol 224, 242–249 (2010).
Ghosh, G. et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest 120, 4141–4154 (2010).
Varambally, S. et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695–1699 (2008).
Strillacci, A. et al. MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res 315, 1439–1447 (2009).
Su, H. et al. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res 69, 1135–1142 (2009).
Wang, H. J. et al. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur J Cancer 46, 2295–2303 (2010).
Thu, K. L., Chari, R., Lockwood, W. W., Lam, S. & Lam, W. L. miR-101 DNA copy loss is a prominent subtype specific event in lung cancer. J Thorac Oncol 6, 1594–1598 (2011).
Semaan, A. et al. MicroRNA-101 inhibits growth of epithelial ovarian cancer by relieving chromatin-mediated transcriptional repression of p21(waf(1)/cip(1)). Pharm Res 28, 3079–3090 (2011).
Wang, R. et al. MiR-101 is involved in human breast carcinogenesis by targeting Stathmin1. PloS One 7, e46173 (2012).
Lin, C., Huang, F., Li, Q. Z. & Zhang, Y. J. miR-101 suppresses tumor proliferation and migration and induces apoptosis by targeting EZH2 in esophageal cancer cells. Int J Clin Exp Pathol 7, 6543–6550 (2014).
Wang, L. et al. miR-101 promotes breast cancer cell apoptosis by targeting Janus kinase 2. Cell Physiol Biochem 34, 413–422 (2014).
Sachdeva, M. et al. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 30, 822–831 (2011).
Cui, T. X. et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 39, 611–621 (2013).
Kim, J. H. et al. Hypoxia-responsive microRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxid Redox Signal. 21, 2469–2482 (2014).
Xu, Y. et al. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep 29, 2019–2024 (2013).
Zheng, F. et al. Systemic delivery of microRNA-101 potently inhibits hepatocellular carcinoma in vivo by repressing multiple targets. PLoS Genet 11, e1004873 (2015).
Bardos, J. I. & Ashcroft, M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta 1755, 107–120 (2005).
Huang, X. et al. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell 35, 856–867 (2009).
Bruning, U. et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol 31, 4087–4096 (2011).
Crosby, M. E., Kulshreshtha, R., Ivan, M. & Glazer, P. M. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res 69, 1221–1229 (2009).
Haque, I. et al. Cysteine-rich 61-connective tissue growth factor-nephroblastoma-overexpressed 5 (CCN5)/Wnt-1-induced signaling protein-2 (WISP-2) regulates microRNA-10b via hypoxia-inducible factor-1alpha-TWIST signaling networks in human breast cancer cells. J Biol Chem 286, 43475–43485 (2011).
Mathieu, J. et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res 71, 4640–4652 (2011).
Neal, C. S., Michael, M. Z., Rawlings, L. H., Van der Hoek, M. B. & Gleadle, J. M. The VHL-dependent regulation of microRNAs in renal cancer. BMC Med 8, 64 (2010).
Loayza-Puch, F. et al. Hypoxia and RAS-signaling pathways converge on and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs. Oncogene 29, 2638–2648 (2010).
Chan, Y. C., Khanna, S., Roy, S. & Sen, C. K. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J Biol Chem 286, 2047–2056 (2011).
Hu, Z. et al. MicroRNA-101 suppresses motility of bladder cancer cells by targeting c-Met. Biochem Biophys Res Commun 435, 82–87 (2013).
Zhang, J. G., Guo, J. F., Liu, D. L., Liu, Q. & Wang, J. J. MicroRNA-101 exerts tumor-suppressive functions in non-small cell lung cancer through directly targeting enhancer of zeste homolog 2. J Thorac Oncol 6, 671–678 (2011).
Jiang, W. et al. miRNA-101 suppresses epithelial-to-mesenchymal transition by targeting HMGA2 in pancreatic cancer cells. Anticancer Agents Med Chem, doi: 10.2174/1871520615666150507122142 (2015).
Koivunen, P. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 (2012).
Shi, Y. et al. Role and mechanism of hypoxia-inducible factor-1 in cell growth and apoptosis of breast cancer cell line MDA-MB-231. Oncol Lett 1, 657–662 (2010).
Wu, D., Jiang, H., Chen, S. & Zhang, H. Inhibition of microRNA-101 attenuates hypoxia/reoxygenationinduced apoptosis through induction of autophagy in H9c2 cardiomyocytes. Mol Med Rep 11, 3988–3994 (2015).
Acknowledgements
This study was supported by grants from SJTU Interdisciplinary Research Grant (YG2013MS36 to HW), National Natural Science Foundation of China (81372233 to HW and 81372188 and 81372188 to JXG and 81402287 to LFL), the State Key Laboratory of Oncogenes and Related Genes in China (JXG, No. 90-14-06), the Special Fund for Innovation and Development of Science and Technology and Cultivation Fund for Major Projects and Innovative Team to JXG, Shanghai Jiao Tong University, China; Tumor Stem Cell Project (#90-14-06 to JXG), Shanghai Cancer Institute, China; The University Doctorate Research Fund for Freshly Recruited Teachers (No. 20130073120010 to LFL), Ministry of National Education, China; the Fund for Key Disciplines and Specialties, Shanghai Health and Family Planning Committee, China (JXG), the Fund for Key Disciplines and Specialties, Shanghai Health and Family Planning Committee, China (JXG) and Startup Funds to JXG from Renji Hospital and School of Medicine, Shanghai Jiao Tong University, China.
Author information
Authors and Affiliations
Contributions
N.L. and H.W. conceived and designed the experiments. N.L., W.Y.X. and H.W. performed the experiments. N.L. and H.W. analyzed the data and wrote the manuscript. W.Y.X., S.S.L., H.Y.C., L.S., M.Y.L., J.X.G., L.F.L., H.M.L., Y.J.F. and P.W. edited the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, N., Xia, WY., Liu, SS. et al. MicroRNA-101 targets von Hippel-Lindau tumor suppressor (VHL) to induce HIF1α mediated apoptosis and cell cycle arrest in normoxia condition. Sci Rep 6, 20489 (2016). https://doi.org/10.1038/srep20489
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20489
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
