
Abstract
Prader-Willi syndrome (PWS) is a neurogenetic disorder caused by loss of paternally expressed genes on chromosome 15q11-q13. The PWS-critical region (PWScr) contains an array of non-protein coding IPW-A exons hosting intronic SNORD116 snoRNA genes. Deletion of PWScr is associated with PWS in humans and growth retardation in mice exhibiting ~15% postnatal lethality in C57BL/6 background. Here we analysed a knock-in mouse containing a 5′HPRT-LoxP-NeoR cassette (5′LoxP) inserted upstream of the PWScr. When the insertion was inherited maternally in a paternal PWScr-deletion mouse model (PWScrp−/m5′LoxP), we observed compensation of growth retardation and postnatal lethality. Genomic methylation pattern and expression of protein-coding genes remained unaltered at the PWS-locus of PWScrp−/m5′LoxP mice. Interestingly, ubiquitous Snord116 and IPW-A exon transcription from the originally silent maternal chromosome was detected. In situ hybridization indicated that PWScrp−/m5′LoxP mice expressed Snord116 in brain areas similar to wild type animals. Our results suggest that the lack of PWScr RNA expression in certain brain areas could be a primary cause of the growth retardation phenotype in mice. We propose that activation of disease-associated genes on imprinted regions could lead to general therapeutic strategies in associated diseases.
Similar content being viewed by others


Introduction
The human SNURF-SNRPN domain located at 15q11q13 (murine 7qC) is subject to genomic imprinting controlled by a bipartite imprinting centre (IC)1 (Fig. 1). This region spans 3–4 Mbp and is considered to be the largest human imprinting cluster identified thus far. Imprinting defects or chromosomal rearrangements/deletions within this locus result in the development of two clinically distinct neurogenetic disorders – the Prader-Willi (PWS) and Angelman (AS) syndromes, respectively.

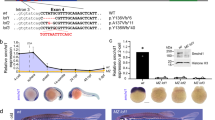
Schematic representation of the PWS-locus in human and mice.
(A) Human chromosome 15q11-q13 region (not drawn to scale). Rectangles or thin ovals denote protein coding gene or snoRNA gene locations and the imprinting center (IC) is shown by a horizontal oval; thin rectangles above the midline denote non-protein coding exons. Arrows indicate promoters and the direction of transcription. The two broken arrows under the top arrow showing the U-UBE3A antisense transcript harboring the two SNORD116 and SNORD115 clusters indicate putative additional primary transcripts with a possible additional promoter upstream from the SNORD115 cluster. (B) Schematic representation of the mouse PWS-locus on chromosome 7; the 5′HPRT-LoxP-NeoR targeting cassette is indicated (not drawn to scale).
AS (MIM 105830) is a severe neurogenetic disorder that is associated with mental retardation, epilepsy, movement disorder and abnormal behaviour. AS is caused by the deletion or inactivating mutations of the UBE3A gene that encodes E3 ubiquitin ligase. In human neurons, UBE3A is exclusively expressed from the maternal chromosome.
PWS (MIM 176270) is a complex neurogenetic disorder with a prevalence of 1 in 10,000 to 1 in 30,000 individuals. Typically, progression of PWS can be subdivided into two main stages: (i) early – characterized by muscular hypotonia resulting in failure to thrive and (ii) late - mostly defined by severe obesity. Additional symptoms common to PWS patients include short stature, small hands and feet, obsessive-compulsive behaviour and mild intellectual disability (for review2,3).
PWS patients lack the paternal expression of imprinted genes located within the PWS locus. Approximately 70% of PWS patients harbour paternally inherited deletions of the entire imprinting domain and approximately 25% of all cases are due to maternal uniparental disomy (two copies of maternal chromosomes)3,4. At most, 3% of PWS patients display genomic imprinting defects3,4.
Several paternally expressed protein-coding genes map to this locus, including NECDIN (NDN), MAGEL2, MKRN3, C15orf2 (recently designated as NPAP1) and the bi-cistronic SNURF-SNRPN (Fig. 1A). Apart from protein-coding genes, this locus also encodes various non-protein-coding RNA (npcRNA) genes; for instance the poorly characterized U-exons and PWRN1/PWRN2 long non-protein-coding transcripts localized between NDN and SNURF-SNRPN genes. In addition, the domain harbours the long U-UBE3A-ATS npcRNA transcript, which spans approximately ~450 kb. It initiates from the U exons upstream of the SNURF/SNRPN gene and extends to the UBE3A gene in antisense orientation (Fig. 1A,B)5. The PWS locus also encodes numerous paternally expressed C/D box snoRNAs. Most of them, if not all are processed from introns of the U-UBE3A-ATS transcript5. This arrangement includes two large tandemly repeated clusters of SNORD116 (HBII-85 snoRNA) and SNORD115 (HBII-52 snoRNA) RNA genes containing 29 and 47 copies, respectively6. Additional snoRNA genes, SNORD107 (HBII-436), SNORD64 (HBII-13) and SNORD108 (HBII-437) are encoded by single and SNORD109A/B (HBII-438a/438b) by two copies5,6 (Fig. 1A). In mouse, the PWS/AS locus maps to chromosome 7C (Fig. 1B). The gene order of the PWS/AS locus and mono-allelic expression in brain are largely conserved. However, in mouse the NPAP1, SNORD108 and SNORD109A/B orthologs are missing, whereas the human locus is devoid of the Frat3 (Peg12) gene6,7 (Fig. 1B).
Analysis of different PWS-locus deletions in mouse models and gene expression in patients with chromosomal translocations predicted the Snord116 gene array to define the PWS critical region (PWScr)8,9,10,11. Previously, we exclusively deleted the Snord116 gene cluster via “chromosome engineering” in mice12. Upon maternal transmission of the deleted allele (PWScrp+/m−), no phenotypic abnormalities were detected. However, if mice were carrying the paternal deletion of the Snord116 cluster (PWScrp−/m+), postnatal growth retardation was observed12. Furthermore, ~15% postnatal lethality was detected in C57BL/6 genetic background, but not in FVB/N or BALB/c12. An independent study eliminating a similar overlapping region in mice, ~45 kb larger than that reported by Skryabin et al. (2007), provided similar results13. In addition, deficiencies in motor learning, increased anxiety, hyperphagia and altered metabolism were reported13. No adolescent obesity was observed in either mouse model12,13. Subsequently, several PWS patients were identified that carry a microdeletion of the SNORD116 gene cluster, strongly supporting that loss of the paternal SNORD116 cluster is responsible for key characteristics of the PWS phenotype14,15,16,17.
In the present study, we analysed a knock-in (KI) mouse model that harbours a 5′HPRT-LoxP-NeoR (hypoxanthine-guanine phosphoribosyltransferase, LoxP and neomycin resistance gene) (5′LoxP) containing cassette ∼27 kb upstream from the Snord116 gene array. We provide biochemical evidence that the insertion does not alter a methylation pattern within the PWS imprinting centre (PWS-IC) and also leaves the expression of protein-coding genes largely unaltered. By contrast, transcription of both the Snord116 and Snord115 gene clusters from the maternal chromosome was observed. We demonstrated that the maternal expression of the Snord116 gene cluster in the PWScrp−/m5′LoxP mouse model rescued both postnatal lethality and growth retardation, which is specifically associated with the PWScrp−/m+ genetic background. Our data emphasized the importance of non-protein-coding RNAs in the etiology of PWS.
Results
Generation of PWScrp−/m5′LoxP mice and growth detection
For the construction of the PWScrp−/m5′LoxP KI mouse model, hypoxanthine-guanine phosphoribosyltransferase (HPRT)-deficient AB2.2 ES cells were modified by homologous recombination (HR) using the targeting construct 5′HPRT/PWScr_targ as the first step in generating the previously reported PWScrp−/m+ mice (Supplementary Fig. 1)12. The construct contained 5′-domains of the HPRT gene controlled by the Pgk promoter, the LoxP site and the neomycin phosphotransferase (Neo) gene, which is transcribed from the promoter of the gene encoding the large subunit of RNA polymerase II (RNAP II) (Fig. 1B, Supplementary Fig. 1C).
The 5′ cassette containing the LoxP site was placed upstream of the Snord116 gene cluster (Fig. 1B Supplementary Fig. 1C).
We obtained a single (from ~250) positive ES clone, which harboured the targeting cassette in the desired location. Expanded ES cells were injected into blastocysts. Chimeras with germ-line transmission were identified by Southern blot analysis as described previously12. Pups of chimeric mice that contained the corresponding 5′LoxP cassette inserted at the desired position, (downstream from Snord64 and upstream from the Snord116 gene cluster) did not reveal discernible phenotypic differences compared to wild type siblings. The heterozygous PWScrp5′LoxP/m+ pups were intercrossed to establish a homozygous KI mouse line. Next, we generated heterozygous KI female mice with modified maternal chromosome (PWScrp+/m5′LoxP) and crossed them with PWScrp−/m+ males to generate PWScrp−/m5′LoxP, PWScrp−/m+ and wild type siblings.
PWScrp−/m5′LoxP pups were slightly, yet distinguishably smaller than their wild-type counterparts when weighed on postnatal day 12 (P12). However, the growth differences became imperceptible after three weeks (P21).
We monitored weight increase of individual PWScrp−/m5′LoxP mice over several weeks in comparison to sibling Snord116 knockout mice (PWScrp−/m+) and wild type (PWScrp+/m+) mice respectively (Fig. 2). The overall weight gain of n = 30 males and n = 26 females of the PWScr116p−/m5′LoxP genotype were compared to n = 30 males and n = 30 females of wild type and n = 26 males and n = 28 females of PWScrp−/m+ animals (Fig. 2; Supplementary Fig. 2; Supplementary Table 1). Analysis of PWScrp−/m5′LoxP growth dynamics revealed a mild growth delay between postnatal day 7 and 19. However, with increasing age the difference vanished and became mostly insignificant in PWScrp−/m5′LoxP P21 males and P22 females compared to wild type mice of identical age (Fig. 2, Supplementary Fig. 2) and no increase in postnatal lethality was detected for PWScrp−/m5′LoxP mice (Supplementary Fig. 3). Hence, measurements and analysis of body weight alteration of PWScrp−/m5′LoxP, PWScrp−/m+ and wild type mice indicated that after the first 3 weeks there was a complete rescue of the growth retardation phenotype in mice harbouring the maternal 5′LoxP cassette compared to PWScrp−/m+ mice with an unmodified maternal chromosome (Fig. 2; Supplementary Table 1). Consistent with our previous results, postnatal growth retardation of PWScrp−/m+ mice was observed beginning on postnatal day 5 in males and on day 6 in females lasting into adulthood (Fig. 2)12.

Growth dynamics of PWScrp−/m5′LoxP, PWScrp−/m+ and male wild type mice.
Curves show the growth dynamics of 86 male mice. The red line shows the weight gain of 30 wild type mice; the green line corresponds to 30 PWScrp−/m5′LoxP mice and the black line corresponds to 26 PWScrp−/m+ mice. Bars indicate standard deviation. Weight mean, observed standard deviation and confidence interval for each time point of the investigated mice, are calculated with a confidence level of 95% (p = 0.05) in Supplementary Table 1.
Expression of genes from PWS/AS locus in mouse models
To delineate the effects of the maternally inherited 5′LoxP cassette at the molecular level, we analysed the expression of non-protein-coding and protein-coding genes within the PWS/AS locus. As mentioned, the mouse PWS locus encodes the Snord64 gene and two large paternally expressed snoRNA clusters: Snord116 and Snord115, respectively. In contrast to human, the expression of the latter two snoRNAs is restricted to the mouse brain1,6,18,19 (Fig. 3).

Expression of PWS/AS locus genes in PWScrp−/m5′LoxP, PWScrp−/m+ and wild type mice.
(A–C) Northern blot analyses of PWS-locus snoRNAs from different tissues of wild type, PWScrp−/m+ and PWScrp−/m5′LoxP mice. Ethidium bromide-stained 5.8 S rRNA is shown as RNA loading control. Tissues and mouse genotypes are indicated on the top of each blot panel. (D) RT-qPCR analysis of PWS/AS-locus genes; the fold change is represented as 2−ΔΔCq. Blue and red bars represent the RNAs expression fold change values of PWScrp−/m+ and PWScrp−/m5′LoxP mice, respectively. The plot represents values of Supplementary Table 2.
We investigated the effects of the 5′LoxP cassette on expression patterns of the distal Snord116 and Snord115 gene clusters. When total RNA from different tissues isolated from 3 male and 3 female mice of wild type, PWScrp−/m5′LoxP and PWScrp−/m+ genotypes were analysed by Northern blot hybridization, we observed ubiquitous RNA expression of both snoRNAs in PWScrp−/m5′LoxP mice (Fig. 3C). In contrast, we did not observe any change in the brain specific expression pattern of Snord64 RNA located upstream of the inserted cassette (Figs 1 and 3A–C). Since expression of Snord115 in PWScrp−/m+ and both snoRNA clusters in wild type mice is restricted to brain (Fig. 3A,B), we conclude that the inserted 5′LoxP cassette results in ubiquitous transcriptional activation of the maternal allele. Brain-specific expression of Snord64 RNA remained unaltered and suggested that only the snoRNA genes located downstream from the transgene insertion were affected (Fig. 3A–C). Interestingly, the observed activation of transcription from the maternal chromosome in PWScrp−/m5′LoxP mice suggests that the transcriptional activation of Snord116 and Snord115 gene clusters is not controlled by the PWS-IC centre and is not maternally silenced.
Next, we performed a comprehensive expression analysis of protein-coding and non-protein-coding genes within the PWS/AS locus using reverse transcription quantitative real-time PCR (RT-qPCR)19,20. Total brain RNA from three male animals of each genotype (wild type, PWScrp−/m5′LoxP and PWScrp−/m+) at postnatal day 7 was extracted independently and RT-qPCR analysis was performed in triplicate for each sample (Fig. 3D, Supplementary Table 2). The mouse PWS – locus harbours several paternally expressed protein-coding genes, including Frat3, Mkrn3, Magel2, Ndn (Necdin) and bi-cistronic Snurf/Snrpn (Fig. 1). Their expression is tightly controlled by the PWS-IC that maps to a ~6 kb region between −3.7 kb and +2.3 kb relative to exon 1 of the mouse Snrpn gene21. Complete or partial deletion of this region results in complete or at least significant loss of gene expression in the PWS locus21,22,23. The RT-qPCR analysis did not reveal significant changes in the corresponding expression levels of all investigated mRNAs within the PWScrp−/m5′LoxP genotypes when compared to both to wild type and PWScrp−/m+ animals, respectively (Fig. 3D, Supplementary Table 2).
Next, we examined the expression profile of PWS-locus encoded non-protein-coding RNAs. Most of them, if not all, are presumed to be processed from the long U-Ube3A-ATS RNA transcript, which initiates from a U-exon on the paternal un-methylated chromosomal region(s), located upstream from the PWS-IC centre (Fig. 1)24. Analogous to the PWS locus protein coding genes, the IC tightly controls expression of the U-Ube3A-ATS pre-RNA and consequently the snoRNAs1,21. In mouse, the transcription of U-Ube3A-ATS pre-RNA is restricted to the neurons of most areas of the brain3. The RNA contains various alternatively spliced exons, which generate numerous large RNAs (identified as ESTs – expressed sequence tags in databases) and snoRNAs. Among the ESTs, there are U-exons, Ipw, Ipw-B – F, repetitive subtypes of Ipw-A and Ipw-G exons, Ube3a cis-antisense transcripts etc. (Fig. 1; Supplementary Fig. 1)6. Imprinted IPW exons A and G flank the Snord116 and Snord115 RNA copies, respectively (Fig. 1)6. To evaluate expression of different PWS locus non-protein-coding transcripts, we have designed primers targeting different U-Ube3a-ATS RNAs and performed RT-qPCR (Supplementary Table 3). Overall, we did not observe any significant differences in the expression of npcRNAs that are located upstream from the 5′LoxP cassette insertion in the PWScrp−/m5′LoxP mice compared to wild type and PWScrp−/m+ animals (Fig. 3D; Supplementary Table 2). The results correlate well with unaltered expression of paternally imprinted protein-coding genes and suggest that regulation by the PWS-IC-centre is not affected (Fig. 3D).
As expected, RT-qPCR analysis of RNA samples extracted from the brains of PWScrp−/m+ mice did not reveal expression of Ipw, IpwA1/A2, Snord116 and IpwB transcripts. In contrast, those RNAs were detected in PWScrp−/m5′LoxP brain samples, once more indicative of maternal gene activation in the KI mice. However, the maternally inherited 5′LoxP cassette led to significantly lower expression values of the aforementioned RNAs when compared to wild type mice (Fig. 3D, Supplementary Table 2). We observed a 7.5 and ~12 fold decrease in the expression levels of Snord116 RNA and IPW transcripts, respectively (Fig. 3D, Supplementary Table 2). In line with our previous observation in PWScrp−/m+ mouse models, the expression of IPW-G exons in PWScrp−/m5′LoxP was also reduced (∼4-fold) in comparison to wild type animals, but the expression levels of Snord115 as detected by RT-qPCR were only slightly lower (∼1.5-fold)12. This could be due to different stabilities of the RNAs. Notably, the expression level of Snord115 in PWScrp−/m+ mice was almost 2-fold lower than in wild type animals, which was not detected in our previous study using Northern blot analysis12. Interestingly, we also observed a slight decrease of Ube3a antisense transcripts that potentially led to a small increase of Ube3a mRNA isoforms expression in PWScrp−/m+ and PWScrp−/m5′LoxP mouse models (Fig. 3D, Supplementary Table 2).
Although we could detect transcription from both RNA polymerase II promoters of the 5′HPRT-LoxP-NeoR cassette, we were not able to conclusively determine the promoter(s) driving expression of the primary transcript harboring the IPW exons and snoRNAs on the maternal chromosome. In any event, our results demonstrate that even a lower expression level of maternal PWScr locus transcription was sufficient to compensate for the growth retardation phenotype associated with the PWScrp−/m+ mice12. Yet, the lower expression level might be the cause of moderate growth retardation observed during the first three weeks of postnatal development.
Maternal inheritance of the 5′LoxP cassette does not affect methylation of the PWS-IC
The imprinted gene expression within the PWS-locus is tightly controlled by the PWS-IC, which is differentially methylated during development. CpG DNA methylation is restricted to the maternal chromosome and causes gene silencing in the locus; the paternal allele, however, remains unmodified and hence transcriptionally active. In mice, the methylation of the PWS-IC is established in oocytes and maintained throughout embryonic development into adulthood1,3. Loss of maternal IC methylation results in the activation of PWS gene expression from the maternal chromosome23. To address possible changes of altered methylation patterns of the PWS-IC in PWScrp−/m5′LoxP mice compared to wild type and PWScrp−/m+ animals, we conducted quantitative real-time PCR (qPCR) analysis. The SacII methylation sensitive restriction endonuclease site in the PWS-IC genomic CpG region was chosen (Fig. 4A). DNA samples from six mice (3 per gender) for each genotype were analysed. Since each round of PCR amplification results in roughly a 2-fold increase in the amount of product, the detected 2-fold differences between SacII digested and intact DNA samples from PWScrp−/m5′LoxP, wild type and PWScrp−/m+ mice indicates that ~50% of the template was cleaved in the endonuclease digested samples (Fig. 4B,C; Supplementary Table 4). Hence, qPCR analysis revealed that CpG methylation of the PWS-IC SacII site on the maternal chromosome was not affected by inheritance of the 5′HPRT-LoxP-NeoR cassette insertion (Fig. 4).

PWS-IC-center CpG methylation analysis.
(A) The mouse PWS-IC genomic region selected for the qPCR assay; cytosine residues that are methylated on the maternal chromosome are shown as capitalized, underlined and italic letters. The SacII endonuclease recognition site is CCGCGG. Quantitative PCR primers are indicated by a black arrows. (B) A summary of the qPCR data is detailed in Supplementary Table 4. For each mouse genotype (WT: wild type; KO: PWScrp−/m+ and 5′ LoxP: PWScrp−/m5′LoxP), six brains were used to isolate DNA samples. Each sample was analysed in triplicate. DNA samples either digested and untreated are indicated by SacII and Uncut respectively. Columns: IC Cq AVE (tech/biol rep) is the average of Cq values obtained from technical and biological replicates per category during the qPCR of the PWS-IC region; IC AVEDEV is the average of standard deviations obtained from all replicates per category during qPCR of the PWS-IC region; Snord64 Cq AVE (tech/biol rep): the average of Cq values obtained during qPCR of the Snord64 gene region; Snord64 AVEDEV: is the respective average standard deviation. (C) Chart visualising 2−ΔΔCq values obtained by qPCR show no differences in CpG methylation of the PWS-IC region among the different mouse genotypes.
Maternal expression of Snord116 in areas of the brain
Since, insertion of the 5′LoxP cassette altered the transcriptional control of PWScr, we investigated the expression of Snord116 in different brain regions of PWScrp−/m5′LoxP mice in comparison to wild type animals. In situ hybridization (ISH) with a Snord116 antisense probe was performed on floating brain sections obtained from six to eight week old mice. The Snord116 distribution between wild type and PWScrp−/m5′LoxP mice was similar (Fig. 5A–C). As negative control, brain sections isolated from PWScrp−/m+ were used. In addition, ISH was performed with a control Snord116 sense probe. No signals were observed in control experiments, even after long exposures (Fig. 5D,E). The strongest ISH signals in brains of both Snord116-expressing mouse lines (wild type and PWScrp−/m5′LoxP) were observed in the hypothalamus, thalamus, hippocampus, anterior olfactory nucleus, piriformal cortex, infralimbic cortex, putamen and dorsal peduncular cortex (Fig. 5). Other regions exhibited moderate Snord116 expression signals. Differences in snoRNA116 expression between wild type and PWScrp−/m5′LoxP mice were noted; for example, in corpus callosum and anterior commissure areas of the brain. The corpus callosum and anterior commissure are bundles of nerve fibers (white matter tracts) involved in interhemispheric communication. They are composed mainly of axons and glial cells. In wild type mice, expression of Snord116 RNA is restricted to neurons, where it is predominantly localised in the nucleolus (with low levels in nucleoplasm)18. In contrast, robust expression of Snord116 in PWScrp−/m5′LoxP mice was observed in non-brain tissue (Fig. 3A) and ISH signals in the corpus callosum and anterior commissure areas of KI mouse brain suggest expression of snoRNA in glial cells (Fig. 5). Nevertheless, although Snord116 was detected in the glial cells of PWScrp−/m5′LoxP mice, the overall brain area patterns of neuronal snoRNA expression between KI and wild type mice was quite similar (Fig. 5, Supplementary Fig. 5).

Snord116 in situ hybridization (ISH) of wild type and PWScrp−/m+5′LoxP mouse sagittal brain sections
(A) Brain sections of wild type mice, the exposure time was one day (B) brain sections of PWScrp−/m+5′LoxP mice. ISH is performed with Snord116 antisense probe. (A,B) Mouse brain arias are denoted as follows: cc: corpus callosum; co: cortex; ha: hypothalamic area; hi: hippocampus; lv: lateral ventricle; on: olfactory nucleus; pu: putamen; ta: thalamic area; pir: piriformal cortex; il: infralimbic cortex; dp: dorsal peduncular cortex. Due to the lower expression levels of Snord116 in the KI mouse, the exposure time had to be increased from 1 to 7 days. (C) Schematic representation of the cuts in sagittal brain sections. (D) Negative control, example of brain sections of PWScrp−/m+ mice hybridized with Snord116 antisense probe. (E) Example of brain sections of wild type mice hybridized with Snord116 sense probe. (D,E) The exposure time was 4 days.
Discussion
Studies of different deletion mouse models and PWS patients have identified a PWS-critical region. This region contains the SNORD116 gene copies flanked by repeated IPW-A exons (elsewhere termed IPW116 or host gene exons – 116HG)12,13,14,15,16,17. Thus far, an important question still remains unanswered. Is the deletion of unknown regulatory elements or lack of non-protein coding RNAs causative of PWS in patients? Recent studies on the activation of PWS-locus gene expression from the maternal chromosome in mice and in PWS-specific induced pluripotent stem cells (iPSCs) indicated the importance of RNAs derived from this region23,25. However, expression of all PWS-locus protein coding and non-protein coding genes was observed and the actual contribution of the Snord116 genes cluster could not be dissected23. Here, based on a KI mouse model we could demonstrate that activating the maternal chromosome region encompassing Snord116 results in rescue of growth retardation and postnatal lethality. The expression levels of Snord116 and IPW-A non-protein coding exons observed in PWScrp−/m5′LoxP mice were considerably lower than those in wild type animals. Although mild growth retardation was observed during the early postnatal period, growth differences between PWScrp−/m5′LoxP and wild type littermates were not discernible after postnatal day 21 and 22 in males and females, respectively. Therefore, our data suggest that expression and to some extent quantity of non-protein coding RNAs play an important role during early postnatal development in mice.
Recently, transcription of the IPW exon containing RNAs had been linked to regulation of maternally expressed genes (MEGs) in the human DLK1-DIO3 imprinted locus in an iPSCs model of PWS. This study suggests that RNA interaction with histone methyltransferase G9A targets the imprinted DNA methylation region (iDMR) in the DLK1-DIO3 locus in trans26. However, the IPW exons and many iDMRs do not show sequence conservation between mammals in general and human and mouse in particular6,27. Therefore, the involvement of IPW-A exons containing long RNA in the regulation of MEGs from Dlk1-Dio3 imprinted domain in mice remains unclear and will be subject to further investigation. Recently, mouse IPW-A exon containing RNA was suggested to regulate diurnal energy expenditure28.
In the PWS critical region, the Snord116 genes exhibit the largest degree of sequence similarity between various mammalian species. Consequently, the focus of investigation was directed to potential snoRNA function. In order to discriminate whether the snoRNA or the long host transcript is responsible for the phenotype, compensation had been attempted by generating Snord116 transgenic mouse lines embedded in introns of different host genes13. A single copy Snord116 gene, inserted within nucleolin intron 11, revealed, as expected, relatively low level of expression13. The mouse lines failed to rescue growth retardation and lethality of the Snrpn to Ube3A deletion mouse model13,29. It is difficult to make any conclusions, as the exact brain localization of the transgene-derived Snord116 was not reported. Our results demonstrate that if Snord116 is responsible for the growth retardation phenotype observed in PWScrp−/m+ mice then ~7.5 fold decrease in Snord116 RNA expression is sufficient to rescue the growth retardation phenotype after postnatal day 21 and 22 in males and females, respectively (Figs 2 and 3D and Supplementary Fig. 2). Importantly, the in situ hybridization experiments revealed that activation of Snord116 expression from the maternal chromosome in PWScrp−/m5′LoxP mice overlaps with the brain areas of wild type mice that express this snoRNA from the paternal chromosome (Fig. 5).
We have preliminary results pertaining to a transgenic mouse line expressing two copies of mouse and one of rat Snord116 processed from introns of a different host gene, namely EGFP (TgSnord116). The expression level of Snord116 observed in the TgSnord116 mouse brain was lower than in wild type animals, but comparable to that in PWScrp−/m5′LoxP mice (Supplementary Fig. 4). However, in contrast to PWScrp−/m5′LoxP, the PWScrp−/m+ TgSnord116 mice once more failed to rescue the growth retardation phenotype. In situ hybridization experiments with a Snord116 - host specific probe revealed that EGFP expression was absent in thalamus, hypothalamus, midbrain and pons of PWScrp−/m+TgSnord116 mice (brain areas where Snord116 is expressed in wild-type; Supplementary Fig. 5). Dysregulation of the hypothalamic endocrine system has been shown to be associated with PWS in humans and could lead to growth retardation in mice30,31. Although, there is a possibility that the host transcript and Snord116 exhibit different stabilities in the aforementioned brain areas, it is conceivable that the absence of Snord116 in the hypothalamus of PWScrp−/m+ TgSnord116 mice explains the lack of compensation for the PWS-like phenotype in these animals.
Thus, the important question of the functional significance of Snord116 C/D box snoRNAs in PWS still needs to be addressed by generating compensatory transgenic animals that express Snord116 in the same brain areas as do wild-type or PWScrp−/m5′LoxP mice. In addition, based on recent findings, the impact of repetitive IPW-A exons containing non-protein coding RNA must be seriously considered.
The present study clearly demonstrates that the lack of expression of non-protein-coding RNAs from the PWS critical region is primarily causative of the growth retardation phenotype in mice. Importantly, the growth retardation observed in PWScrp−/m+ animals could be rescued by the transcriptional activation of the PWScr region from the silenced maternal chromosome. Our results suggest that activation of disease-associated genes on imprinted regions could lead to general therapeutic strategies in man. In fact, recent findings that topoisomerase inhibitors, such as topotecan, could activate the silent paternal Ube3a allele in neurons support this notion32,33.
Material and Methods
Generation of transgenic mice
Details on the 5′-LoxP targeting cassette construction were previously given12. In brief, two mouse genomic fragments generated from the Snord116 upstream region were PCR amplified from a PAC clone (RPCIP711K19517Q6, RZPD German Resource Centre for Genome Investigation). These fragments were used as homologues arms in the targeting vector flanking the region containing the 5′-portion of the hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene, the LoxP site and the neomycin resistance gene (Fig. 1B and Supplementary Fig. 1C)12. The resulting DNA construct was linearized with NotI endonuclease and electroporated at 25 μF and 400 V (gene Pulser; Bio-Rad) into AB2.2 embryonic stem (ES) cells (kindly provided by A. Bradley) resuspended in buffer containing 20 mM HEPES pH 7.4, 173 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 6 mM dextrose and 0.1 mM ß-mercaptoethanol. ES cells, passage 17, were grown in HEPES-buffered, Dulbecco’s modified Eagle’s medium supplemented with 15% fetal bovine serum (HyClone), nonessential amino acids, L-glutamine, ß-mercaptoethanol, 1000 U/ml recombinant LIF (Chemicon) and antibiotics (penicillin 100 U/ml and streptomycin 100 μg/ml) on a g-irradiated monolayer of primary fibroblast feeder cells12. Positive ES clones were selected using a nested PCR approach and Southern blot hybridization with a 32P-labled 5′HR probe12. One KI ES clone containing an inserted 5′LoxP cassette was injected into 3.5-day-old B6D2F1 (C57BL/6 × DBA) blastocysts and the resulting embryos were transferred to CD-1 foster mice. Chimeras were identified by their agouti coat color.
Southern blot analysis
Positively targeted ES cell clones or mouse tail biopsies were analyzed by Southern-blotting. Approximately 5 μg of genomic DNA was digested with EcoRI (or EcoRV), fractionated on 0.8% agarose gels and transferred to GeneScreen nylon membranes (NEN DuPont). The membranes were hybridized with a 32P-labeled 1.7-kb probe containing sequences 5′ to the targeted homology and washed with (final concentrations) 0.5x SSPE (1 × SSPE is 0.18 M NaCl, 10 mM NaH2PO4 and 1 mM EDTA [pH 7.7]) and 0.5% sodium dodecyl sulfate at 65 °C.
Northern blot hybridization
Five μg of total RNA samples isolated from different organs of PWScrp−/m5′LoxP, PWScrp−/m+ and wild type mice were separated on 8% (w/v) (acrylamid/N,N′-bis-acrylamide (29:1)) denaturing polyacrylamide gels (PAAG; 7 M urea, 1 × TBE buffer) and transferred onto positively charged nylon membranes (BrightStar Plus, Ambion or Hybond-N+, Amersham Biosciences) using a Trans-blot semi-dry blotting apparatus (BioRad) at 400 mA for 45 min in 0.5 × TBE buffer (90 mM Tris, 64.6 mM boric acid, 2.5 mM EDTA, pH 8.3). Membranes were first pre-hybridized for 30 min in 0.5 M sodium phosphate (pH 6.5), 7% (w/v) sodium dodecyl sulfate (SDS) buffer at 56 °C followed by overnight hybridization at the same temperature. Northern blot hybridization was performed with 5′-32P-labelled snoRNA specific oligonucleotides that were also used for reverse transcription in RT-qPCR assays (see below) (Supplementary Table 3). Membranes were washed twice at 46 °C in 2 × SSC buffer (20 mM sodium phosphate, 0.3 M NaCl, 2 mM EDTA, pH 7.4) containing 0.5% SDS for 30 min (washing was repeated when the blot showed high counts). Blots were exposed to MS-film (Kodak) for a few hours at −80 °C, if necessary, exposure time was extended overnight at −80 °C.
RT qPCR analysis
Reverse transcription quantitative real-time PCR analysis was performed as previously reported19,20. Briefly, total RNA was isolated from mouse brains using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA samples, 5 μg each, were treated with RNase-free DNase I (Roche), followed by reverse transcription with 0.5 μl of oligo(dT)12–18 (500 ng/μl) and 1 μl of random hexamer primer (3 μg/μl) oligonucleotide mix. All qPCR reactions were performed in triplicate, in a total volume of 10 μl containing 2 μl of cDNA (~20 ng), 5 μl of 2 × LightCycler 480 SYBR Green Master Mix (Roche) and 1 μM of each primer (Supplementary Table 3). The amplification program was as follows: 5 min initial denaturation step at 95 °C, with subsequent 45 cycles of 20 sec at 95 °C and 1 min at 60 °C. The reaction included single acquisition of fluorescent signal at 60 °C for each cycle and continuous acquisition from 50 °C to 97 °C at the end of the 45 cycles for melt-curve analysis. Quantification Cycle (Cq) values were calculated using Light-cycler 480 SW 1.5 software (Roche) and all data were transferred to Excel files for subsequent analysis. Data analysis was performed using a geometric mean of ActB mRNA and U1 snRNA selected as reference genes. The fold change is represented as 2−ΔΔCq (Supplementary Table 2). The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines were followed in all qPCR experiments34.
PWS-IC qPCR methylation analysis
Genomic DNA was isolated from mouse brains using the proteinase K and phenol/chloroform extraction method. Five μg of each methyl-sensitive endonuclease digestion DNA samples were incubated with 20 units of SacII (Roche) restriction enzyme overnight at 37 °C. Quantitative real-time PCR reactions were performed in triplicate in a total volume of 10 μl containing 2 μl of DNA (1:50 dilution of SacII digested or untreated DNA samples), 5 μl of 2 × LightCycler 480 SYBR Green Master Mix (Roche) and 1 μM of each primer. The amplification program and data acquisition was carried out as described above. Data analysis was performed using Snord64 as reference control. Fold change is represented as 2−ΔΔCq (Supplementary Table 4).
In situ hybridization
Mice were transcardially perfused with 0.1 M phosphate buffered saline (PBS, pH 7.2), followed by freshly prepared 4% paraformaldehyde in PBS (PFA). The brains were removed and fixed overnight in 4% PFA35. In situ hybridization was performed on floating 30 μm brain sections as previously described36. The Snord116 probe was synthesized in vitro and cloned into the pUC minusMCS plasmid, custom made by Blue Heron Biotech, LLC. The plasmid DNA was linearized with either BamHI (for antisense probe) or SalI (for sense probe) and transcribed in vitro in the presence of S35 α-UTP using T7 and T3 RNA polymerases, respectively. The ISH was performed at 50 °C overnight with the corresponding RNA probes ~4.5 × 106 cpm/ml. The sections were washed once with 2X SSC and 50% deionized formamide buffer for 10 min at 50 °C followed by a single 10 min wash in 2X SSC at the same temperature and incubated with RNase A (90 ng/ml) for 45 min at 40 °C. Subsequently, the sections were washed by gentle stirring: first in 5 L of 2X SSC buffer for 45 min at 50 °C, then in 5 L of 0,1X SSC, 0,05% sodium pyrophosphate, 14 mM β-mercaptoethanol solution for 3 h at 50 °C, followed by a slow cooling of the buffer to room temperature and continued washing overnight. The slices were mounted on Superfrost Gold Plus (Menzel) microscopy glass slides, dried and exposed to autoradiography films (Kodak Biomax MR).
Mice
All mouse procedures were performed in compliance with the guidelines for the welfare of experimental animals issued by the Federal Government of Germany and approved by the State Agency for Nature, Environment and Consumer Protection North Rhine-Westphalia (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen). Animals were kept in specific pathogen-free animal facilities. All breeding was conducted in a controlled (21 °C, 30–50% humidity) room with a 12:12 hour light-dark cycle. Mice were housed under non-enriched, standard conditions in individually ventilated (36 (l) × 20 (w) × 20 (h) cm) cages for up to five littermates. Pups were weaned 19–23 days after birth and females were kept separately from males. Body weight statistic analysis was performed as previously described by Skryabin et al. 200712.
Additional Information
How to cite this article: Rozhdestvensky, T. S. et al. Maternal transcription of non-protein coding RNAs from the PWS-critical region rescues growth retardation in mice. Sci. Rep. 6, 20398; doi: 10.1038/srep20398 (2016).
References
Horsthemke, B. & Buiting, K. Genomic imprinting and imprinting defects in humans. Adv Genet 61, 225–246, doi: 10.1016/S0065-2660(07)00008-9 (2008).
Butler, M. G. Prader-Willi Syndrome: Obesity due to Genomic Imprinting. Curr Genomics 12, 204–215, doi: 10.2174/138920211795677877 (2011).
Cassidy, S. B., Schwartz, S., Miller, J. L. & Driscoll, D. J. Prader-Willi syndrome. Genet Med 14, 10–26, doi: 10.1038/gim.0b013e31822bead0 (2012).
Buiting, K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 154C, 365–376, doi: 10.1002/ajmg.c.30273 (2010).
Runte, M. et al. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet 10, 2687–2700 (2001).
Cavaille, J. et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci USA 97, 14311–14316 (2000).
Relkovic, D. & Isles, A. R. Behavioural and cognitive profiles of mouse models for Prader-Willi syndrome. Brain Res Bull 92, 41–48, doi: 10.1016/j.brainresbull.2011.09.009 (2013).
Ding, F. et al. Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models. Mamm Genome 16, 424–431 (2005).
Gallagher, R. C., Pils, B., Albalwi, M. & Francke, U. Evidence for the role of PWCR1/HBII-85 C/D box small nucleolar RNAs in Prader-Willi syndrome. Am J Hum Genet 71, 669–678 (2002).
Runte, M., Varon, R., Horn, D., Horsthemke, B. & Buiting, K. Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader-Willi syndrome. Hum Genet 116, 228–230, doi: 10.1007/s00439-004-1219-2 (2005).
Schule, B. et al. Molecular breakpoint cloning and gene expression studies of a novel translocation t(4;15)(q27;q11.2) associated with Prader-Willi syndrome. BMC Med Genet 6, 18, doi: 10.1186/1471-2350-6-18 (2005).
Skryabin, B. V. et al. Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet 3, e235 (2007).
Ding, F. et al. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PloS one 3, e1709, doi: 10.1371/journal.pone.0001709 (2008).
Bieth, E. et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet: EJHG 23, 252–255, doi: 10.1038/ejhg.2014.103 (2015).
de Smith, A. J. et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet 18, 3257–3265, doi: 10.1093/hmg/ddp263 (2009).
Duker, A. L. et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet: EJHG 18, 1196–1201, doi: 10.1038/ejhg.2010.102 (2010).
Sahoo, T. et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 40, 719–721 (2008).
Vitali, P., Royo, H., Marty, V., Bortolin-Cavaille, M. L. & Cavaille, J. Long nuclear-retained non-coding RNAs and allele-specific higher-order chromatin organization at imprinted snoRNA gene arrays. J Cell Sci 123, 70–83, doi: 10.1242/jcs.054957 (2010).
Galiveti, C. R., Raabe, C. A., Konthur, Z. & Rozhdestvensky, T. S. Differential regulation of non-protein coding RNAs from Prader-Willi Syndrome locus. Sci Rep 4, 6445, doi: 10.1038/srep06445 (2014).
Galiveti, C. R., Rozhdestvensky, T. S., Brosius, J., Lehrach, H. & Konthur, Z. Application of housekeeping npcRNAs for quantitative expression analysis of human transcriptome by real-time PCR. RNA 16, 450–461, doi: 10.1261/rna.1755810 (2010).
Dubose, A. J., Smith, E. Y., Yang, T. P., Johnstone, K. A. & Resnick, J. L. A new deletion refines the boundaries of the murine Prader-Willi syndrome imprinting center. Hum Mol Genet 20, 3461–3466, doi: 10.1093/hmg/ddr262 (2011).
Bressler, J. et al. The SNRPN promoter is not required for genomic imprinting of the Prader-Willi/Angelman domain in mice. Nature genetics 28, 232–240, doi: 10.1038/90067 (2001).
Wu, M. Y., Jiang, M., Zhai, X., Beaudet, A. L. & Wu, R. C. An unexpected function of the Prader-Willi syndrome imprinting center in maternal imprinting in mice. PloS one 7, e34348, doi: 10.1371/journal.pone.0034348 (2012).
Landers, M. et al. Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic acids research 32, 3480–3492, doi: 10.1093/nar/gkh670 (2004).
Cruvinel, E. et al. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader-Willi syndrome iPSCs. Human molecular genetics 23, 4674–4685, doi: 10.1093/hmg/ddu187 (2014).
Stelzer, Y., Sagi, I., Yanuka, O., Eiges, R. & Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat Genet 46, 551–557, doi: 10.1038/ng.2968 (2014).
Stelzer, Y. et al. Identification of novel imprinted differentially methylated regions by global analysis of human-parthenogenetic-induced pluripotent stem cells. Stem Cell Reports 1, 79–89, doi: 10.1016/j.stemcr.2013.03.005 (2013).
Powell, W. T. et al. A Prader-Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum Mol Genet 22, 4318–4328, doi: 10.1093/hmg/ddt281 (2013).
Tsai, T. F., Jiang, Y. H., Bressler, J., Armstrong, D. & Beaudet, A. L. Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum Mol Genet 8, 1357–1364 (1999).
Swaab, D. F. Prader-Willi syndrome and the hypothalamus. Acta Paediatr Suppl 423, 50–54 (1997).
Zhang, Q., Bouma, G. J., McClellan, K. & Tobet, S. Hypothalamic expression of snoRNA Snord116 is consistent with a link to the hyperphagia and obesity symptoms of Prader-Willi syndrome. Int J Dev Neurosci 30, 479–485, doi: 10.1016/j.ijdevneu.2012.05.005 (2012).
Huang, H. S. et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481, 185–189, doi: 10.1038/nature10726 (2012).
Powell, W. T. et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc Natl Acad Sci USA 110, 13938–13943, doi: 10.1073/pnas.1305426110 (2013).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–622, doi: 10.1373/clinchem.2008.112797 (2009).
Khanam, T. et al. Two primate-specific small non-protein-coding RNAs in transgenic mice: neuronal expression, subcellular localization and binding partners. Nucleic Acids Res 35, 529–539, doi: 10.1093/nar/gkl1082 (2007).
Tiedge, H. The use of UV light as a cross-linking agent for cells and tissue sections in in situ hybridization. DNA Cell Biol 10, 143–147 (1991).
Acknowledgements
This work was supported by the National Genome Research Network (grant #NGFNIII 01GS0808) and by the Interdisciplinary Centre of Clinical Research (IZKF), Muenster.
Author information
Authors and Affiliations
Contributions
T.S.R., J.B. and B.V.S. conceived and designed the study. T.R. performed ISH experiments, C.R.G. completed qPCR analyses. C.A.R. did the computational work. B.S., L.V.G. and B.V.S. generated KI mice and performed growth analysis. A.W. was involved in Northern blot analysis. T.S.R. and B.V.S. analysed data and wrote the manuscript. All authors provided input and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Rozhdestvensky, T., Robeck, T., Galiveti, C. et al. Maternal transcription of non-protein coding RNAs from the PWS-critical region rescues growth retardation in mice. Sci Rep 6, 20398 (2016). https://doi.org/10.1038/srep20398
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20398
This article is cited by
-
Ectopic expression of Snord115 in choroid plexus interferes with editing but not splicing of 5-Ht2c receptor pre-mRNA in mice
Scientific Reports (2019)
-
Neuronal differentiation induces SNORD115 expression and is accompanied by post-transcriptional changes of serotonin receptor 2c mRNA
Scientific Reports (2018)
-
BC1 RNA motifs required for dendritic transport in vivo
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
