
Abstract
Progranulin (PGRN) is a recently identified adipokine that is supposed to have anti-inflammatory actions. The proinflammatory cytokine interleukin-1β (IL1β) stimulates several mediators of cartilage degradation. Toll like receptor-4 (TLR4) can bind to various damage-associated molecular patterns, leading to inflammatory condition. So far, no data exist of PGRN effects in inflammatory conditions induced by IL1β or lipopolysaccharide (LPS). Here, we investigated the anti-inflammatory potential of PGRN in IL1β- or LPS-induced inflammatory responses of chondrocytes. Human osteoarthritic chondrocytes and ATDC-5 cells were treated with PGRN in presence or not of IL1β or LPS. First, we showed that recombinant PGRN had no effects on cell viability. We present evidence that PGRN expression was increased during the differentiation of ATDC-5 cell line. Moreover, PGRN mRNA and protein expression is increased in cartilage, synovial and infrapatellar fat pad tissue samples from OA patients. PGRN mRNA levels are upregulated under TNFα and IL1β stimulation. Our data showed that PGRN is able to significantly counteract the IL1β-induced expression of NOS2, COX2, MMP13 and VCAM-1. LPS-induced expression of NOS2 is also decreased by PGRN. These effects are mediated, at least in part, through TNFR1. Taken together, our results suggest that PGRN has a clear anti-inflammatory function.
Similar content being viewed by others


Introduction
Osteoarthritis (OA) is a multifactorial joint degenerative disease characterized by progressive destruction of articular cartilage, changes in subchondral bone, osteophyte formation and synovial inflammation. It is the most prevalent type of arthritis, but its aetiology is still largely unknown1. Although OA is commonly described as non-inflammatory disease, inflammation is recognized as contributing to the symptoms and progression of OA2. On the other side, obesity is an important risk factor for OA that may result in overloading of joints and by a chronic ‘low-grade inflammatory systemic state sustained by a dysregulation of adipokines in white adipose tissue and other peripheral tissues, including joint tissues, that can contribute to an altered immune and inflammatory response3,4. Several adipokines can also be produced by chondrocytes and act locally in cartilage homeostasis5,6.
Progranulin (PGRN) is a recently identified adipokine7, also known as GP88, acrogranin, proepithelin, granulin/epithelin precursor (GEP) or PC cell-derived growth factor (PCDGF). PGRN is a 68–88 kDa secreted glycoprotein that is produced by a broad range of tissues, including human articular cartilage8 and adipose tissue7. PGRN has been implicated in a wide variety of biological functions, including wound healing9, bone regeneration10 and inflammation11,12. It has been previously reported that PGRN levels were significantly elevated in cartilage of patients with OA and rheumatoid arthritis (RA)8. Moreover, serum levels of PGRN in RA patients were found to be significantly higher than those in age-matched healthy controls13. Recently, it has been demonstrated that PGRN levels are increased at local site of inflammation and are associated to disease activity in patients with RA14. PGRN also plays a crucial role in chondrocyte proliferation15, differentiation and endochondral ossification of growth plate during development16,17. In addition, the group of Chuanju Liu reported that PGRN antagonised tumour necrosis factor α (TNF-α) through binding to TNF receptors (TNFR) and exhibited an anti-inflammatory function, by suppressing the pro-inflammatory action of TNF-α in a arthritis murine models18,19. Additionally, these authors found that deficiency of PGRN led to spontaneous OA-like phenotype in ‘aged’ mice. PGRN-deficient mice exhibited breakdown of cartilage structure, while local delivery of recombinant PGRN protein attenuated degradation of cartilage matrix in surgically induced OA models. Furthermore, PGRN plays a protective role by promoting anabolism of degenerative chondrocytes mainly through TNF receptor 2 (TNFR2). TNF receptor 1 (TNFR1) binding to PGRN prevents the activation of the NF-κB pathway by TNFα, which induces MMPs and ADAMTS, thus inhibiting cartilage degradation20.
Secreted pro-inflammatory cytokines, such as Interleukin 1β (IL1β), are critical mediators implicated in OA pathophysiology, which make them primary targets for therapeutic strategies21. IL1β is released by synoviocytes, chondrocytes and invading macrophages in inflamed joints and it is well-established that IL1β plays a pivotal role in the pathogenesis of OA22. IL1β triggers a cascade of cartilage damage events like the production of more pro-inflammatory cytokines, the synthesis of catabolic factors and the release of some inflammatory mediators such as prostaglandins produced by COX-2 and nitric oxide (NO) up-regulated by NOS223. Moreover, it has been suggested that IL1β induces the expression of the TNF-α gene in chondrocytes24 and upregulates the surface expression of TNFRs25.
Given that innate immune responses are important in the development of OA, the role of Toll like receptors (TLR) have been recently taken into account26. Among TLRs, TLR4 has been involved in OA27. TLR4 activation by lipopolysaccharide (LPS) involves the production of NO28, several pro-inflammatory cytokines29 and adipokines5,30 that may work together boosting cartilage degradation31. Previously, Yin et al. reported that PGRN-deficient macrophages challenged with LPS increase pro-inflammatory cytokine production32 and Hwang et al. showed that PGRN efficiently inhibited LPS-mediated pro-inflammatory signalling in endothelial cells through attenuation of the NF-κB pathway, suggesting its beneficial anti-inflammatory effects33.
To the best of our knowledge, no data exist about PGRN effect on IL1β- or LPS-induced inflammatory responses of chondrocytes. Therefore, we investigated the effect of PGRN on the expression of some inflammatory mediators and catabolic factors in IL1β- or LPS-stimulated chondrocytes for a better understanding of the underlying mechanisms implicated.
Materials and Methods
For experiments involving humans, all the methods were carried out in accordance with the approved guidelines. All experimental protocols were approved by the local ethics committee (SERGAS, Santiago University Clinical Hospital Ethics Committee (CAEIG Comité Autonómico de Ética da Investigación de Galicia 2014/310). Informed consent was obtained from all subjects.
Reagents
Fetal bovine serum (FBS), MTT dye, human transferrin, sodium selenite, ethylenediaminetetraacetic acid (EDTA) solution, mouse recombinant TNF-α, mouse and human recombinant IL1β and lipopolysaccharide (LPS) were obtained from Sigma (St. Louis MO, USA). Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F12 medium, l-glutamine, antibiotics and trypsin-EDTA were purchased from Lonza (Verviers, Belgium). Recombinant mouse PGRN (18–589) was purchased from Enzo Life Sciences (NYC, USA) and recombinant human PGRN (1–593) was from AdipoGen (Switzerland).
Cell culture and treatments
The murine chondrogenic cell line ATDC-5 (purchased from RIKEN Cell Bank, Tsukuba, Japan) was cultured in DMEM–Ham’s F-12 medium supplemented with 5% fetal bovine serum, 10 μg/ml human transferrin, 3·10−8 M sodium selenite, l-glutamine and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin). Chondrogenic ATDC-5 cells were differentiated into mature chondrocytes. Briefly, cells were seeded at a density of 6·104/well in 6-well plates with the ATDC-5 standard media supplemented with insulin (10 mg/mL). The differentiation media was replaced every two days for 14 days. Differentiation was qualitatively characterized by increased formation of cell nodules. In other experiments (data not shown), differentiation was further analyzed by sequential increase in the levels of type II collagen, aggrecan and type X collagen mRNA, as previously published34. The immortalized human juvenile costal chondrocyte cell line T/C-28a2 (a kind gift from Dr. M.B. Goldring, Hospital for Special Surgery, NYC, USA) was culture in DMEM–Ham’s F-12 medium supplemented with 10% fetal bovine serum, l-glutamine and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin). Primary chondrocytes were harvested from human OA articular cartilage samples obtained from articular joints of patients undergoing total knee replacement surgery (with permission from the local ethics committee (SERGAS, Santiago University Clinical Hospital Ethics Committee (CAEIG Comité Autonómico de Ética da Investigación de Galicia 2014/310) and informed consent was obtained from all patients participating in the study) as previously described35. Human chondrocytes were cultured in DMEM/Ham’s F12 medium supplemented with 10% of fetal bovine serum, L-glutamine and antibiotics (50 units/ml penicillin and 50 mg/ml streptomycin). Cells were seeded in monolayer up to the high density and used in the first passage of culture in order to avoid dedifferentiation.
For RT-PCR and western blot, cells were seeded in 6-well plates until complete adhesion and then incubated overnight in serum-free conditions. Then, cells were treated as indicated in each case.
Cell viability
Cell viability was tested using the methyl-thiazolyl-tetrazolium (MTT) reagent to detect functional mitochondria in living cells. Briefly, ATDC-5, ATDC-5 mature and T/C-28a2 cells (8·103/well) were seeded in 96-well plates. After overnight starvation, they were incubated with recombinant mouse and human PGRN for 48 h in serum free medium. Doses of 100 and 200 ng/ml of PGRN reflect serum concentrations in healthy individuals36,37. Then, cells were incubated for 4 h with MTT reagent. After formazan salt was dissolved, absorbance was measured at 550 nm using a microtiter enzyme-linked immunosorbent assay reader (Multiskan EX; Thermo Labsystems). Cells cultured without PGRN were used to normalize cell viability status.
Nitrite assay
Nitrite accumulation was determined in the culture medium using the Griess reaction, as previously described31. Briefly, 100 μl cell culture medium was mixed with 100 μl Griess reagent (equal volumes of 1% [weight/vol] sulfanilamide in 5% [vol/vol] phosphoric acid and 0.1% [weight/vol] naphtylethylenediamine-HCl), incubated at room temperature for 10 min and then the absorbance at 550 nm was measured using a microplate reader (Titertek-Multiscan, Labsystem, Helsinki, Finland). Fresh culture medium was used as blank in all of the experiments. The amount of nitrite in the samples (in micromolar units) was calculated from a sodium nitrite standard curve freshly prepared in culture medium.
RNA Isolation and Real-time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Mouse and human PGRN mRNA levels were determined using SYBR Green–based quantitative PCR. RNA was extracted using a NucleoSpin kit (Macherey-Nagel), according to the manufacturer’s instructions. For relative quantification, we performed an RT reaction using 1 mg of RNA with a Thermo Scientific Verso cDNA Synthesis Kit (42 °C for 30 min, followed by incubation at 95 °C for 2 min). RT-PCR was performed in a Stratagene MX3005P thermal cycler using a standard protocol (95 °C for 10 min followed by 40 cycles for 15 s of denaturation at 95 °C and 1 min annealing/extension at 60 °C), a SABiosciences Master Mix and specific primers (for mouse GAPDH, 140 bp, PPM02946E, reference position 309, GenBank accession no. NM_008084.2; for human GAPDH, 175 bp, PPH00150E, reference position 1287, GenBank accession no. NM_002046.3; for mouse PGRN, 121 bp, PPM24687A, reference position 1704, GenBank accession no. NM_008175.4; for human PGRN, 130 bp, PPH02482B, reference position 1975, GenBank accession no. NM_002087.2; for mouse NOS2, 122 bp, PPM02928B, reference position 2728–2748, GenBank accession no. NM_010927.3; for mouse TNFRSF1A, 156 bp, PPM03087D, reference position 1461, GenBank accession no. NM_011609.4; for mouse TNFα, 110 bp, PPM03113G, reference position 749, GenBank accession no. NM_013693). Results of comparative RT-PCRs were analyzed using MxPro software (Stratagene, CA, USA).
Protein extraction and Western blot analysis
After the cell treatment, cells were rapidly washed with ice-cold phosphate buffered saline and scraped in lysis buffer for protein extraction (10 mM Tris/HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 30 mM Sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 0.5% Triton X-100, 1 mM PMSF and protease inhibitor cocktail from Thermo Scientific). Lysed cells were centrifuged at 14.000 g for 20 min. SDS-PAGE and blotting procedure were carried on as previously described38. Immunoblots were incubated with the appropriate antibody (anti-PGRN diluted 1:1000, Santa Cruz, CA, USA; anti-NOS2 diluted 1:1000, Cell Signalling, MA, USA; anti-COX2 diluted 1:50, anti-VCAM-1 diluted 1:1000, Cell Signalling, MA, USA; anti-MMP13 diluted 1:500, Santa Cruz, CA, USA) and visualized with an Immobilon Western Detection kit (Millipore, MA) using anti-rabbit (GE Healthcare, UK) horseradish-peroxidise-labelled secondary antibody diluted 1:2000. To confirm equal loading in each sample, the membranes were stripped in stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl pH 6.7) and re-blotted with anti-GAPDH antibody diluted 1:30000 (Sigma, MO, USA). The images were captured and analyzed with an EC3 imaging system (UVP). Densitrometric analyses were performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
siRNA-mediated gene silencing of TNFα receptor
In order to silence TNFR gene, we used the siRNA that targets TNFRSF1A (Integrated DNA Technologies, USA) and the siRNA negative control, that does not target any known sequence. Transfection with 10 nM of siRNA duplex was performed using the cationic lipid siLentFect (BioRad, CA, USA) according to the manufacturer’s recommendations. Cells were transfected for 48 h and then they were stimulated with 0.1 ng/ml of IL1β in presence or not of 200 ng/ml of PGRN for 48 h.
Statistical analysis
Data are reported as the mean ± standard error of the mean (SEM) and followed normal distribution of independent experiments, which were done at least three times, each time with at least three independent observations. Statistical analysis was performed using two-tailed Student’s t test and one-way ANOVA test followed by the Bonferroni’s test for multiple-comparisons using the Prism computerized package (GraphPad Software V.5, La Jolla, CA, USA). P-values less than 0.05 were considered significant.
Ethics approval
This study was conducted with the approval of the Santiago University Clinical Hospital Ethics Committee (CAEIG 2014/310).
Results
Effect of PGRN on cell viability
First, in order to assess the effect on vitality of PGRN, we performed MTT assay in undifferentiated ATDC-5, in mature ATDC-5 and human T/C-28a2 chondrocytes. Treatment with increasing concentrations of PGRN for 48 hours showed no toxic effect on cell vitality (Sup. Fig. 1).
PGRN mRNA and protein production during ATDC-5 differentiation
To determine whether the levels of PGRN mRNA and protein change during chondrocyte differentiation, we differentiated ATDC-5 cells into mature and hypertrophic chondrocytes. As shown in Fig. 1, PGRN mRNA expression increased along differentiation process of ATDC-5 cells. This increase is highly significant after 7, 14 and 21 days of differentiation in comparison to undifferentiated cells (Day 0) (Fig. 1, upper panel). This effect was observed also at protein expression (Fig. 1, lower panel).

PGRN mRNA and protein expression during ATDC-5 differentiation after 7, 14 and 21 days.
Values are the mean ± SEM of at least 4 independent experiments (**P < 0.01 vs. Ctrl, ***P < 0.001 vs. Ctrl.). Cell lysates underwent Western blot analysis using PGRN antibody. GAPDH was used as a loading control. The blots were run under the same experimental conditions. The full blots are shown in Supplementary Fig. 2. Blots are representative of at least 3 independent experiments.
PGRN expression in osteoarthritic and healthy tissues
Since local production of adipokines may have important pathological implications on cartilage homeostasis, we also assessed the expression of PGRN mRNA and protein levels in human primary chondrocytes, infrapatellar fat pad (IPFP) and synovium of OA patients and compare this expression between OA subjects and healthy donors. As shown in Fig. 2, human primary chondrocytes, IPFP and synovium expressed efficiently PGRN. Moreover, PGRN expression was significantly higher in chondrocytes, IPFP and synovial tissues obtained from OA patients compared to those obtained from healthy donors.

Determination of PGRN mRNA and protein in healthy and osteoarthritis human tissues.
PCR results were shown in fold change, where grey bars represent the PGRN mRNA expression in chondrocytes, infrapatellar fat pads (IPFPs) and synovial tissues obtained from osteoarthritis (OA) patients (*P < 0.05 vs. Ctrl.). PGRN protein expression was showed by representative western blots of 18 OA patients (age 52–73; mean BMI 28.4) and 6 healthy donors (age 23–50; mean BMI 23.4). GAPDH was used as loading control. Western blots have been run under the same experimental conditions. The full blots are shown in Supplementary Fig. 3.
Effect of pro-inflammatory cytokines and TLR-4 agonist on PGRN mRNA expression in ATDC5 differentiated cells
To elucidate the pattern of PGRN expression under pro-inflammatory conditions, we treated differentiated ATDC-5 (14 days) cells with different doses of TNFα, IL1β, IL6 and LPS as a TLR-4 agonist. These differentiated cells have a phenotype of adult mature chondrocytes and, in spite of its murine origin, have an identical behaviour to human cells, at last in terms of inflammatory mediators expression39. As shown in Fig. 3a, cells stimulated with TNFα during 24 h showed a marked increase in PGRN mRNA expression at 1 and 5 ng/ml. A similar effect was observed when cells were stimulated with IL1β (0.05 and 0.1 ng/ml; Fig. 3b) during 24 h. Nonetheless, neither IL6 nor LPS had significant effects on PGRN expression (Fig. 3c,d).

Mouse PGRN mRNA expression after TNFα, IL1β, IL6 and LPS treatment.
(a) Cells were treated with TNFα 0.1, 1, 5 ng/ml, (b) IL1β 0.025, 0.05, 0.1 ng/ml, (c) IL6 0.1, 0.5, 1 ng/ml and (d) LPS 100, 250, 500 ng/ml for 24 h. Values are the mean ± SEM of at least 3 independent experiments (**P < 0.01 vs. Ctrl, ***P < 0.001 vs. Ctrl.).
PGRN decreased IL1β induction of chondrocyte catabolism
In order to gain further insights into the potential pharmacological activity of PGRN, human primary chondrocytes were treated with IL1β (10 ng/ml) in presence or not of PGRN 200 ng/ml for 48 h. As shown in Fig. 4a, human primary chondrocytes challenged with IL1β, showed a strong accumulation of NO after 48 h in comparison to control unstimulated cells. However, treatment with recombinant PGRN significantly (P < 0.01) led to inhibition of IL1β-induced NO production, whereas PGRN per se did not affect basal NO production. Nitrite accumulation inhibition was further confirmed by western blot analysis of NOS type 2 (NOS2). PGRN was able to clearly inhibit NOS2 protein expression (Fig. 4b). Furthermore, since PGRN is supposed to prevent cartilage destruction in inflammatory arthritis mediated by TNF-α, we examined the levels of catabolic biomarkers COX-2 and MMP13 in human primary chondrocytes challenged with IL1β. COX-2 and MMP13 protein levels were significantly increased after 48 h of IL1β treatment. However, PGRN significantly decreased IL1β-induced COX-2 and MMP13 expression (Fig. 4c,d). Moreover, since pro-inflammatory cytokines such as IL1β and TNFα are able to up-regulate VCAM-1 expression in primary cultures of human articular chondrocytes, we examined VCAM-1 protein levels after PGRN treatment. IL1β is able to induce VCAM-1 expression. The treatment with PGRN produced a significant decrease of IL1β-induced VCAM-1 expression (Fig. 4e).

PGRN suppressed IL1-β induction of chondrocyte catabolism.
(a) Cells were treated with 10 ng/ml of IL1β, in presence or absence of 200 ng/ml of PGRN for 48 h. Culture medium was subsequently analyzed for nitrite levels. NO concentration (μM) was determined using the Griess reaction. Values are the mean ± SEM of at least 3 independent experiments (**P < 0.01 vs. IL1β) (a, upper panel). Cell lysates underwent Western blotting analysis under the same condition (a, lower panel). After transferring the blots onto PVDF membranes, we cropped the targeted blots according to referenced indicating markers and then targeted proteins were immunoblotted with NOS2, COX-2, MMP13 and VCAM-1 antibodies. GAPDH was used as a loading control. Blots are representative of at least 3 independent experiments. (b–e) Western blot densitometric analysis (n = 3; ***P < 0.001 PGRN + IL1β vs. IL1β).
PGRN counteracted IL1β and LPS-driven NOS2 through TNFR1
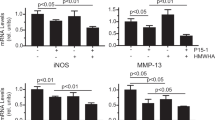
It was reported that PGRN can competitively bind to TNFR1 and TNFR2 and prevents TNFα-mediated inflammation18. On the basis of above introduced results showing that PGRN counteracts IL1β-driven inflammatory response, we sought to investigate whether this anti-inflammatory effect of PGRN was mediated through TNFR. Firstly, we corroborated that PGRN was able to modulate IL1β or LPS-induced NO levels and NOS2 expression also in differentiated ATDC5 cells (Fig. 5a,b). Then, to determine whether the anti-inflammatory effect of PGRN was mediated through TNFR, we analysed the role of TNFR1 using a siRNA approach. Mature ATDC5 cells were transfected with siTNFR1 and then treated with IL1β or LPS in presence or not of PGRN (Fig. 5c,d). As shown in Fig. 5, TNFR1 siRNA knockdown supressed the anti-inflammatory effect of PGRN under IL1β (panel c) or LPS (panel d) stimulation.

PGRN counteracted IL1β and LPS-induced nitric oxide (NO) production and inhibited NOS2 expression in cultured ATDC-5 cells, at least in part, through TNFR1.
(a,b) Cells were treated with 0.1 ng/ml of IL1β (a) and 250 ng/ml of LPS (b) alone or in combination PGRN (200 ng/ml) during 48 h. Culture medium was subsequently analyzed for nitrite levels. NO concentration (μM) was determined using the Griess reaction. Values are the mean ± SEM of at least 3 independent experiments (a) **P < 0.01 PGRN + IL1β vs. IL1β; (b) **P < 0.01 PGRN + LPS vs. LPS). Cell lysates underwent Western blotting analysis using NOS2 antibody. GAPDH was used as a loading control. The blots were run under the same experimental conditions. The full blots are shown in Supplementary Fig. 3. Blots are representative of at least 3 independent experiments. (c,d) Cells were transfected with negative control siRNA or TNFR1-targeted siRNA (siTNFR1) and stimulated with 0.1 ng/ml of IL1β (c) and 250 ng/ml of LPS (d) in absence or presence of PGRN 200 ng/ml for 48 h. Relative mRNA levels of NOS2 were measure by RT-PCR. Values are the mean ± SEM of at least 3 independent experiments (c) **P < 0.01 siC- PGRN + IL1β vs. siC- IL1β; (d) **P < 0.01 siC- PGRN + LPS vs. siC-LPS).
Discussion
The recently discovered adipokine PGRN has been matter of extensive studies in the last few years. The role of PGRN in inflammation has been only partially defined40. Although PGRN is one of the major anti-inflammatory molecules, it is possible that PGRN has dual roles in inflammation and might exert pro- or anti-inflammatory functions depending on different tissues. For instance, PGRN inhibits LPS-mediated IL-6, TNF-α and MCP-1 cytokine release from macrophages41, but PGRN is also the major adipokine involved in high-fat diet-induced insulin resistance by inducing up-regulation of IL-6 expression, a pro-inflammatory cytokine7. Chronic inflammation in RA is mainly mediated by TNF-α42. Recently, PGRN was recognized as a novel ligand of TNFR1/2. Actually, PGRN blocks TNF-α-mediated signalling pathways by competing with TNF-α binding to receptors in inflammatory arthritis murine models18,19.
However, there are currently no approaches in the literature evaluating PGRN action neither through IL1β nor through TLR4 activation in chondrocytes. Previously, Yin et al. reported that PGRN-deficient macrophages challenged with LPS increase pro-inflammatory cytokine production32 and Hwang et al. showed that PGRN efficiently inhibited LPS-mediated pro-inflammatory signalling in endothelial cells through attenuation of the NF-κB pathway, suggesting its beneficial anti-inflammatory effects33. Thus, we have studied the effect of PGRN on the expression of some inflammatory mediators and catabolic factors in IL1β- or LPS-stimulated chondrocytes. To the best of our knowledge, this is the first experimental evidence that PGRN is able to reduce mediators of inflammation induced by IL1β or LPS in human chondrocytes.
Here we show for the first time, that PGRN mRNA and protein expression is detected in human infrapatellar fat pads (IPFPs) and synovial tissues. Moreover, PGRN expression was significantly higher in chondrocytes, IPFP and synovial tissues obtained from OA patients compared to these tissues obtained from healthy donors. Our findings are in agreement with previous results of increased PGRN mRNA and protein expression in OA and RA cartilage described by Guo et al.8 Recently, it was reported the expression profile of PGRN in another cartilage degenerative disease, such as disc degenetation. PGRN levels were also elevated in intervertebral discs during aging and PGRN knockout mice developed an early onset of degeneration in intervertebral cartilage43.
Our work makes another novel observation, that PGRN mRNA expression was increased during the process of ATDC-5 cell differentiation into mature chondrocytes. These data suggested that this adipokine might play a role in the complex mechanisms of chondrocyte development and differentiation. PGRN might be a marker of late-phase chondrogenic differentiation. To this regard, the group of Chuan-ju Liu reported that PGRN was a key downstream molecule of BMP2 and it was required for BMP2-mediated chondrocyte differentiation through Erk1/2 signaling17. They also showed that PGRN was required for BMP-2 induction of osteoblastogenesis and ectopic bone formation10. Moreover, we observed elevated PGRN levels also in hypertrophic chondrocytes, those localized above subchondral bone. However, in PGRN −/− mice, Zhao et al. revealed osteophyte formation and ectopic subchondal sclerosis. These results indicated disorder of bone metabolism in cartilage and subchondral bone of these mice, suggesting a role for PGRN in the regulation of subchondral bone turnover20.
Although several suppositions postulated that PGRN might be strongly influenced by the inflammatory “milieu”, here we show clear experimental evidence that classic cytokines or activation of TLR4 by LPS in chondrocytes are able to strongly influence PGRN levels. These findings suggest that PGRN induction by TNFα and IL1β released from joint tissues may be an adaptive response to the inflammatory state.
Since IL1β plays a pivotal role in the pathogenesis of OA22, counteracting its pro-inflammatory effects with PGRN might be an interesting therapeutic strategy. Actually, we assessed whether PGRN was able to inhibit IL1β-mediated pro-inflammatory response. Our data clearly demonstrate that PGRN strongly attenuated IL1β driven inflammatory markers, such as NO production, NOS2, COX2 and VCAM-1 expression, as well as catabolic markers of cartilage breakdown, such as MMP13. Moreover, we showed clear evidence that PGRN effect upon IL1β-driven NOS2 induction is mediated, at least in part, through TNFR1. Actually, by down-regulating TNFR1 expression through RNA interference, the anti-inflammatory role of PGRN counteracting IL1β was totally blocked, suggesting that PGRN is working via TNFα signalling through binding to TNFR.
Here we also show clear evidence that PGRN is able also to modulate LPS-driven NOS2 induction. This effect is likely to be mediated, at least in part, through TNFR1. Actually, by down-regulating TNFR1 expression through siRNA, the anti-inflammatory role of PGRN was totally blocked. Our results are in line with other published results suggesting that PGRN is able to inhibit LPS-mediated pro-inflammatory signalling32,33. Thus, at the functional aspect, the activity of PGRN and its increased expression observed upon IL-1β and TNF-alpha treatment could mediate and sustain clear pro-anabolic and anti-inflammatory pathways. Nonetheless, the relevance of these data in the pathophysiology of osteoarthritis or other chronic rheumatic diseases should be supported by clinical data that at present are non-existent.
In conclusion, on the basis of our data and on previous published results, PGRN in articular joint cells could limit expression of inflammatory mediators, adhesion molecules and enzymes involved in cartilage breakdown triggered by cytokines and LPS that ultimately compromise cell survival and tissue function. PGRN may represent a novel key molecule coupling cartilage metabolism with inflammation to switch physiologic and clinical phenotypes. Thus, targeted strategies aimed to increase PGRN levels appeals to be an attractive therapeutic strategy for OA, which could be potentially achieved through multiple ways including life style modifications and pharmacological approaches.
Additional Information
How to cite this article: Abella, V. et al. The novel adipokine progranulin counteracts IL-1 and TLR4-driven inflammatory response in human and murine chondrocytes via TNFR1. Sci. Rep. 6, 20356; doi: 10.1038/srep20356 (2016).
References
Goldring, M. B. & Goldring, S. R. Osteoarthritis. J Cell Physiol 213, 626–634 (2007).
Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage 21, 16–21 (2013).
Gómez, R. et al. What’s new in our understanding of the role of adipokines in rheumatic diseases? Nat Rev Rheumatol 7, 528–36 (2011).
Lago, F., Dieguez, C., Gómez-Reino, J. & Gualillo, O. Adipokines as emerging mediators of immune response and inflammation. Nat Clin Pract Rheumatol 3, 716–24 (2007).
Conde, J. et al. Expanding the adipokine network in cartilage: identification and regulation of novel factors in human and murine chondrocytes. Ann Rheum Dis 70, 551–559 (2011).
Conde, J. et al. Adipokines: biofactors from white adipose tissue. A complex hub among inflammation, metabolism and immunity. BioFactors 37, 413–420 (2011).
Matsubara, T. et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab 15, 38–50 (2012).
Guo, F. et al. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum 62, 2023–2036 (2010).
Zhao, Y. P., Tian, Q. Y. & Liu, C. J. Progranulin deficiency exaggerates, whereas progranulin-derived Atsttrin attenuates, severity of dermatitis in mice. FEBS Lett 587, 1805–1810 (2013).
Zhao, Y. P., Tian, Q. Y., Frenkel, S. & Liu, C. J. The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling. Biomaterials 34, 6412–6421 (2013).
Wu, H. & Siegel, R. M. Medicine. Progranulin resolves inflammation. Science 332, 427–428 (2011).
Jian, J., Konopka, J. & Liu, C. Insights into the role of progranulin in immunity, infection and inflammation. J Leukoc Biol 93, 199–208 (2012).
Yamamoto, Y. et al. Increased Serum GP88 (Progranulin) Concentrations in Rheumatoid Arthritis. Inflammation 88, (2014).
Cerezo, L. A. et al. Progranulin Is Associated with Disease Activity in Patients with Rheumatoid Arthritis. Mediators Inflam 2015, 740357 (2015).
Xu, K. et al. Cartilage oligomeric matrix protein associates with Granulin-Epithelin Precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J Biol Chem 282, 11347–11355 (2007).
Bai, X.-H. et al. ADAMTS-7, a direct target of PTHrP, adversely regulates endochondral bone growth by associating with and inactivating GEP growth factor. Mol Cell Biol 29, 4201–4219 (2009).
Feng, J. Q. et al. Granulin epithelin precursor: a bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J 24, 1879–1892 (2010).
Tang, W. et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484 (2011).
Tian, Q. et al. Three TNFR-binding domains of PGRN act independently in inhibition of TNF-alpha binding and activity. Front Biosci 19, 1176–1185 (2014).
Zhao, Y. -p. et al. Progranulin protects against osteoarthritis through interacting with TNF- alpha and beta-Catenin signalling. Ann Rheum Dis (2014). 10.1136/annrheumdis-2014-205779
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J.-P. & Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 7, 33–42 (2011).
Goldring, S. R. & Goldring, M. B. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res S27–S36 (2004). 10.1097/01.blo.0000144854.66565.8f
Goldring, M. B. & Berenbaum, F. The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res S37–S46 (2004). 10.1097/01.blo.0000144484.69656.e4
Campo, G. M. et al. Hyaluronan in part mediates IL-1beta-induced inflammation in mouse chondrocytes by up-regulating CD44 receptors. Gene 494, 24–35 (2012).
Saperstein, S., Chen, L., Oakes, D., Pryhuber, G. & Finkelstein, J. IL-1beta augments TNF-alpha-mediated inflammatory responses from lung epithelial cells. J Interferon Cytokine Res 29, 273–84 (2009).
Gómez, R., Villalvilla, A., Largo, R., Gualillo, O. & Herrero-Beaumont, G. TLR4 signalling in osteoarthritis—finding targets for candidate DMOADs. Nat Rev Rheumatol (2014). 10.1038/nrrheum.2014.209
Wang, P., Zhu, F., Tong, Z. & Konstantopoulos, K. Response of chondrocytes to shear stress: antagonistic effects of the binding partners Toll-like receptor 4 and caveolin-1. FASEB J 25, 3401–3415 (2011).
Clancy, R. M., Gomez, P. F. & Abramson, S. B. Nitric oxide sustains nuclear factor kappaB activation in cytokine-stimulated chondrocytes. Osteoarthritis Cartilage 12, 552–558 (2004).
Abdollahi-Roodsaz, S. et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest 118, 205–216 (2008).
Otero, M. et al. Phosphatidylinositol 3-kinase, MEK-1 and p38 mediate leptin/interferon-gamma synergistic NOS type II induction in chondrocytes. Life Sci 81, 1452–60 (2007).
Otero, M., Lago, R., Lago, F., Reino, J. J. G. & Gualillo, O. Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: synergistic effect of leptin with interleukin-1. Arthritis Res Ther 7, 581–91 (2005).
Yin, F. et al. Exaggerated inflammation, impaired host defense and neuropathology in progranulin-deficient mice. J Exp Med 207, 117–128 (2010).
Hwang, H. J. et al. Progranulin Protects Vascular Endothelium against Atherosclerotic Inflammatory Reaction via Akt/eNOS and Nuclear Factor-κB Pathways. PLoS ONE 8, (2013).
Gómez, R., Lago, F., Gómez-Reino, J. J., Dieguez, C. & Gualillo, O. Expression and modulation of ghrelin O-acyltransferase in cultured chondrocytes. Arthritis Rheum 60, 1704–9 (2009).
Conde, J. et al. Adiponectin and leptin induce VCAM-1 expression in human and murine chondrocytes. PloS ONE 7, e52533 (2012).
Li, H. et al. Circulating PGRN is Significantly Associated with Systemic Insulin sensitivity and Autophagic Activity in Metabolic Syndrome. Endocrinology en20141058 (2014). 10.1210/en.2014-1058
Youn, B. S. et al. Serum progranulin concentrations may be associated with macrophage infiltration into omental adipose tissue. Diabetes 58, 627–636 (2009).
Lago, R. et al. A new player in cartilage homeostasis: adiponectin induces nitric oxide synthase type II and pro-inflammatory cytokines in chondrocytes. Osteoarthritis Cartilage 16, 1101–9 (2008).
Santoro, A. et al. Choosing the right chondrocyte cell line: Focus on nitric oxide. J Orthop Res (2015). 10.1002/jor.22954
D’Acquisto, F., Rattazzi, L., Piras, G. & Galuppo, M. L. Novel immunological targets in rheumatic diseases: clues from current therapies. Drug Discov Today 19, 1155–60 (2014).
Yin, F. et al. Exaggerated inflammation, impaired host defense and neuropathology in progranulin-deficient mice. J Exp Med 207, 117–128 (2010).
Matsuno, H. et al. The role of TNF-alpha in the pathogenesis of inflammation and joint destruction in rheumatoid arthritis (RA): a study using a human RA/SCID mouse chimera. Rheumatology 41, 329–337 (2002).
Zhao, Y. et al. Progranulin knockout accelerates intervertebral disc degeneration in aging mice. Sci Rep 5, 9102 (2015).
Acknowledgements
Oreste Gualillo and Francisca Lago are Staff Personnel of Xunta de Galicia (SERGAS) through a research-staff stabilization contract (ISCIII/SERGAS). OG is a member of RETICS Programme, RD12/0009/0008 (RIER: Red de Investigación en Inflamación y Enfermedades Reumáticas) via FEDER and Instituto de Salud Carlos III (ISCIII). The OG work was funded by Instituto de Salud Carlos III and FEDER (grants PI14/00016 and PIE13/00024). Vanessa Abella is a recipient of a predoctoral fellowship from ESF (European Social Fund) grant from Xunta de Galicia through a contract signed with University of Coruña. Morena Scotece is recipient of the “FPU” Program of the Spanish Ministry of Education. Javier Conde is a recipient of a fellowship from the Foundation IDIS-Ramón Dominguez. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
V.A. participated in acquisition of data, analysis and interpretation of data and critical revision of the manuscript. M.S., J.C., C.P., V.L., J.P., R.G., F.L. and M.Á.G.-G. participated in acquisition of data and samples, drafting of the manuscript and statistical analysis. O.G. participated in conception and design of the study, in analysis and interpretation of data, critical revision of the manuscript and scientific supervision of experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Abella, V., Scotece, M., Conde, J. et al. The novel adipokine progranulin counteracts IL-1 and TLR4-driven inflammatory response in human and murine chondrocytes via TNFR1. Sci Rep 6, 20356 (2016). https://doi.org/10.1038/srep20356
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20356
This article is cited by
-
Adipokines, adiposity, and atherosclerosis
Cellular and Molecular Life Sciences (2022)
-
Progranulin signaling in sepsis, community-acquired bacterial pneumonia and COVID-19: a comparative, observational study
Intensive Care Medicine Experimental (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
