
Abstract
We report two methods of surface enhanced Raman spectroscopy (SERS) for hemozoin detection in malaria infected human blood. In the first method, silver nanoparticles were synthesized separately and then mixed with lysed blood; while in the second method, silver nanoparticles were synthesized directly inside the parasites of Plasmodium falciparum. It was observed that the first method yields a smaller variation in SERS measurements and stronger correlation between the estimated contribution of hemozoin and the parasitemia level, which is preferred for the quantification of the parasitemia level. In contrast, the second method yields a higher sensitivity to a low parasitemia level thus could be more effective in the early malaria diagnosis to determine whether a given blood sample is positive.
Similar content being viewed by others
Introduction
Malaria is a global disease which causes 584,000 deaths per year in the world1. Malaria parasites in infected erythrocytes metabolize hemoglobin resulting in the production of hemozoin which can serve as a unique biomarker2. Early diagnosis is important in malaria infection management and greatly enhances clinical outcome. Currently microscopic examination of Giemsa-stained blood smears is regarded as the “gold standard” in malaria diagnosis. However, the process is time consuming and requires trained operators to ensure parasite detection especially at low parasitemia levels3. Alternative diagnostic techniques that do not require trained personal, are fast and cheap, require minimum sample preparation and are equally sensitive compared to microscopy would greatly aid the global effort of malaria elimination, especially in those regions with low resources.
Numerous approaches have been explored to develop diagnostic tools that are more reliable and/or sensitive than the current gold standard. These include rapid diagnostic tools (RDTs)4, cell dyn machine5, magneto optical technology (MOT) using polarized light6, laser desorption mass spectrometry (LDMS)7, magnetic resonance8, polymerase chain reaction (PCR)9,10, quantitative buffy coat method11, flow cytometry12, serological tests13, optical tweezer14 and attenuated total reflectance infrared spectroscopy (ATR-IR)15. A number of these methods are suitable to be applied in low resource clinical settings but often do not have the desired level of sensitivity needed. These include RDT, CDM, QBC, MOT and a few single cell measurement methods as described below. All of the approaches show reduced sensitivity or alternatively specificity or both particularly at low parasitemia levels. The sensitivity and specificity of RDT can be over 90%, but the accuracy will decrease significantly when the parasite density is lower than 500 parasites/μl4 and in addition it is difficult to use RDT to quantify the parasitemia level4. The sensitivity values of cell dyn machine (CDM) and quantitative buffy coat (QBC) methods are only 50% (for parasitemia levels less than 0.1%) and 33.3% (for parasitemia levels around 0.02%), respectively. MOT using polarized light requires a strong magnetic field while the sensitivity and specificity are only 26.7% and 80.6%, respectively, in a field test on 46 patient showing uncomplicated malaria infection detected by clinical diagnosis16. Flow cytometry can only measure a parasitemia level of 0.1% or higher in culture12.
Alternative approaches that can achieve high sensitivity at low parasitemia levels are predominately not suitable for field use due to high cost or low throughput. Laser desorption mass spectrometry is highly sensitive but is expensive and requires extensive sample preparation7, while serological tests have high sensitivity and specificity (91% and 86%, respectively) but are time consuming and require expertise in sample interpretation13. Similarly approaches using optical tweezers are very time consuming and are therefore not suitable to scan a large volume of samples14. Confocal Raman spectroscopy has been conducted to observe hemozoin crystals in the vacuoles of parasites but again its throughput is low17. Conventional MRI offers a non-invasive method to demonstrate pathological features of cerebral malaria18 but its operation cost is high. To overcome the high cost and bulky equipment limitations of conventional MRI, a micromagnetic resonance relaxometry (MRR) method using a simple low cost and portable MRI device has been reported19. MRR can achieve fast and quantitative measurements of the parasitemia level as low as 0.0002% for P. falciparum infection but the interference of hemoglobin may affect the accuracy of MRR19. PCR is sufficiently sensitive to detect cells infected by a single parasite and track the stage change during parasite development in real time9. Importantly it can differentiate between different species of human malaria parasites20. However, PCR still has to be seen as time-consuming and sample preparation is still relatively complicated even with the recently developed LAMP based isothermal PCR21. ATR-IR has even achieved a sensitivity comparable to PCR15. This technique can detect blood infected by parasites in the ring stage with a parasitemia level at 0.00001% and it has been also used to quantify the parasitemia level. However, the actual sensitivity in quantification is only 0.1% and the standard deviation of the estimated parasitemia level is as high as 0.05%. Moreover, it requires an expensive diamond crystal as the sample stage.
Raman spectroscopy has been used to characterize the electronic structure of β-hematin (a biocrystal with close Raman characteristics resemblance to hemozoin) or hemozoin22 and its change with drug treatment or disease progression23. Moreover, spontaneous Raman spectroscopy combined with other approaches such as confocal microscopy24 or optical tweezers25 have shown great promise for malaria diagnosis26. However, the amount of hemozoin produced in the early ring stages normally seen in peripheral blood is very small and parasites younger than 6 hours post invasion do not contain hemozoin detectable by the dark-field microscopy method27, even dark-field microscopy has improved sensitivity as compared to traditional light microscopy28. Therefore, the enhancement of Raman signals is crucial for the detection of hemozoin in early ring stages so as to provide the sensitivity needed for diagnosis17.
Surface enhanced Raman spectroscopy (SERS) has been reported to enhance the Raman signal of hemozoin or β-hematin by several orders of magnitude29. In tip-enhanced Raman spectroscopy (TERS)30, an atomic force microscope tip covered with SERS-active silver nanoparticles has been shown to enhance the Raman signal originated from hemozoin in the parasite vacuoles by 106 to 107 folds. The enhancement of Raman signals occurs when hemozoin crystals are in close proximity to or direct contact with noble metal surfaces. Further enhancement can be achieved by surface enhanced Raman resonance scattering (SERRS) when there is close Raman shift match between the excitation laser wavelength and hemozoin electronic transition31. This dramatic enhancement of Raman signal suggests the potential use of SERS in early malaria diagnosis where the parasitemia levels and hemozoin concentration in blood samples are very low. However, like other techniques focusing on single cell characterization17,22,25,32, the performance of SERS on a single blood cell highly depends on the precision of locating the cell, which can be very challenging at a low parasitemia level. While theoretically possible, to the best of our knowledge SERS has never been used to quantify the parasitemia level of malaria infected blood samples for early malaria diagnosis. Furthermore, the lowest detection limit of SERS in the current literature is about 0.0005%, which was achieved using a gold coated substrate based on butterfly wings33. However, this method requires the development of a cost-effective and reproducible method to fabricate complex butterfly wing nanostructures. Moreover, Raman measurements have to be taken from specific locations where the aggregation of hemozoin yields maximum enhancement. This makes it time consuming at a low parasitemia level, since hemozoin spreads sparsely on the substrate when the parasitemia level is low.
We have previously used magnetic field enriched SERS to augment the interaction between β-hematin and silver nanoparticles31,34 enabling a detection limit as low as 5-nM β-hematin. However, the requirement of an external magnetic field is inconvenient, which could induce additional variation in SERS readings. Here, we report two enhanced SERS methods for the detection of hemozoin from Plasmodium falciparum infected human blood. In Method 1, silver nanoparticles are synthesized separately and then mixed with the processed sample just as conducted in other SERS studies35,36. While in Method 2, silver nanoparticles are synthesized directly inside parasites to achieve closer contact with hemozoin, which is a new technique enabling ultrasensitive detection of hemozoin. Our methods are unique in that it can potentially achieve both cost effectiveness and high sensitivity when combined with the development of a cost effective Raman spectrometer37 for spectral measurements and paper based microfluidic chips38 for sample preparation. The detection limit achieved by our methods in terms of parasitemia level is comparable to those most sensitive techniques including MRR, PCR and the conventional Giemsa stained microscopy; meanwhile its cost will be only slightly higher than those of RDT.
Results
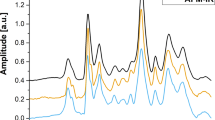
Figure 1 shows (a) the SERS spectra and (b) hemozoin contribution as a function of parasitemia level for infected and normal blood samples obtained using Method 1. Prominent vibrational features of hemozoin, such as ν15 at 754 cm−1, ν2 at 1570 cm−1 and ν(C=C) at 1623 cm−1 are shown in most of the spectra in Fig. 1(a). Detailed peak assignments are shown in Supplementary Table 1. Hemozoin contribution refers to the weight of the basic component spectrum corresponding to hemozoin resulting from the least square regression as described in data processing section. The lowest detectable parasitemia level was determined to be 0.01% after conducting a series of t-tests to compare the hemozoin contribution in infected blood samples with that in the normal blood sample (p < 0.05). There appears to be a strong correlation between hemozoin contribution and the parasitemia level in Fig. 1(b). The second-order polynomial relation between the SERS intensity and parasitemia level could be explained as follows. Since the volume of silver nanoparticles in this study was fixed, there would be fewer and fewer silver nanoparticles for hemozoin to bind when the concentration of hemozoin increased with the parasitemia level. So the SERS intensity increased linearly with parasitemia level at the beginning but then level off, which looked like a parabola.

SERS spectra and hemozoin contribution in Method 1.
(a) SERS spectra contributed by hemozoin in infected blood treated by Method 1. (b) Hemozoin contribution as a function of parasitemia level. In (a), the data point corresponding to a parasitemia level of 0.01% is marked by a red box. In (b), the resulting curve of the second-order polynomial fitting for the data points corresponding to parasitemia levels in the range of 0.01% to 0.2% is shown in red. The data corresponding to normal blood samples, labeled as “NB”, is added manually to facilitate comparison. The y axes in both figures are plotted in arbitrary units, labeled as “A. U.” The asterisks indicate parasitemia levels at which the Raman peak intensity at 1623 cm−1 were significantly different from that in the normal blood sample in t-test (p < 0.05). The detection limit was determined to be 0.01% parasitemia level (marked by the red box) in this manner.
Figure 2 shows the SERS spectra and ν10 peak intensity as a function of parasitemia level obtained using Method 2, in which silver nanoparticles were synthesized inside parasites. Compared with the spectra in Fig. 1, the Raman spectra in Fig. 2 are much stronger. Prominent vibrational features of hemozoin, such as ν15 at 754 cm−1, ν2 at 1570 cm−1 and ν(C=C) at 1623 cm−1 are shown in all spectra. Detailed peak assignments are shown in Supplementary Table 1. Multiple hemozoin Raman peaks are visible in the spectra of normal blood samples, which can be attributed to hematin converted from hemoglobin during sample preparation. In Method 2, most hemoglobin were filtered out, whereas a tiny amount could still stay in or attached to parasites. During silver nanoparticles synthesis, the use of detergent Triton X-100 and hydroxylamine hydrochloride likely converted the residue hemoglobin to heme and then hematin39,40 which contributed to the Raman peaks41. Note that hematin and hemozoin share most peaks including 1623 cm−1. The detection limit for Method 2 is determined to be 0.00005% parasitemia level in the ring stage, after comparing the Raman peak intensity at 1623 cm−1 measured from infected blood samples with that from the normal blood sample with t-test (p < 0.001). The detection limit is exceptionally low compared to most relevant publications we can find (shown in Table 1).

SERS spectra and hemozoin Raman peak distribution in Method 2.
(a) SERS spectra of normal blood and infected blood sample treated by Method 2. (b) SERS peak intensity at 1623 cm−1 as a function of parasitemia level. The data corresponding to normal blood samples, labeled as “NB”, is added manually to facilitate comparison. The y axes in both figures are plotted in arbitrary units, labeled as “A. U.” The asterisks indicate parasitemia levels at which the Raman peak intensity at 1623 cm−1 were significantly different from that in the normal blood sample in t-test (p < 0.001). The detection limit was determined to be 0.00005% parasitemia level (marked by the red box) in this manner.
To illustrate the difference in the separation of parasites and nanoparticles between Methods 1 and 2, Fig. 3 shows the Giemsa stained images of a typical infected blood sample, silver nanoparticles and a blood sample treated by Method 2 before and after nanoparticle synthesis as well as a blood sample treated by Method 1 for comparison. Figure 3(a) illustrates the purple rings superimposed on top of red blood cells, which are the stained DNAs of malaria parasites in the ring stage. Figure 3(b) displays the Giemsa stained image of silver nanoparticles serving as a reference to facilitate search for nanoparticles in subsequent images, in which aggregated nanoparticles show up as sparsely distributed brown or black dots. In Fig. 3(c), red blood cells have been lysed as treated by Method 2 so the blood cells do not exist in background, which is visibly different from Fig. 3(a). However, parasites were not lysed because the concentration of the lysing agent was purposefully reduced. For this reason, the stained DNA rings are clearly seen. In Fig 3(d), nanoparticles have been synthesized in the lysed blood sample as treated by Method 2. It can be clearly seen that some nanoparticles are in the close proximity to the DNA rings of parasites as indicated by red arrows. This phenomenon can be observed in roughly one out of every ten rings in samples treated by Method 2. This suggests that silver nanoparticles were more likely to be close to hemozoin inside the same parasites. Because hemozoin was not released to the outside of parasites, the original local hemozoin concentrations in the parasites were maintained, which created higher SERS signals than otherwise. For the purpose of comparison, Fig. 3(e) illustrates the Giemsa stained image of a blood sample treated by Method 1, in which lysed blood samples and parasites were mixed with nanoparticles separately synthesized. Because parasites have been lysed, no DNA rings were observed and hemozoin inside parasites were thus released into the entire sample. Hemozoin concentration in the sample was thus much lower than that in the original parasites. The SERS signal from hemozoin was thus much lower as expected.

Giemsa stained images of blood samples treated by Method 1 and Method 2.
(a) a blood sample with parasites in the ring stage prior to cell lysis, (b) silver nanoparticles alone, (c) a blood sample after blood cell membrane lysis but prior to nanoparticle synthesis treated by Method 2, (d) the blood sample after nanoparticle synthesis treated by Method 2 and (e) a blood sample treated by Method 1 and then mixed with nanoparticles synthesized separately.
Discussion
Asymptomatic patients with extremely low levels of parasitemia pose a key challenge in any effort to control malaria. The extremely low levels of circulating parasites in the peripheral blood42 make accurate diagnosis particularly difficult. Here we present data that clearly demonstrates the feasibility and advantage of surface enhanced Raman spectroscopy for detecting malaria parasites under those circumstances. While the lowest detectable parasitemia level for Method 1 is approximately 0.01% ring stage parasites, comparable to other methods such as QBC and RDTs in malaria diagnosis (Table 1), Method 2 has a detection limit as low as 0.00005%, which equates to approximately 2.5 parasites/μl of blood and is comparable to the most sensitive detection techniques currently available. The strong correlation between hemozoin concentration and parasitemia level seen in Method 1 has the additional advantage of allowing the direct quantification of the parasite level, which would be of great value for the rapid determination of the overall severity of malaria infection. The relatively high ratio of standard deviation to mean seen at each parasitemia level in Method 1 (15% to 58%) is likely the result of variations in contaminating cell debris that adhere to hemozoin crystals and silver nanoparticles during the centrifugation step. This contamination would directly affect the interaction between hemozoin and silver nanoparticles. In addition the lysis method using Triton-X and sonication may not be able to release all hemozoin out of every parasite’s vacuole leading to an overall reduced signal although cost effective. Future work will focus on improving the lysis step to further increase the sensitivity of Method 1.
The exceptionally high sensitivity of Method 2 is better than most existing methods (Table 1). The close proximity of nanoparticles to hemozoin concentrated inside parasites achieved in this approach (Fig. 3) dramatically enhances the Raman signal of hemozoin leading to the improved detection limit. While very promising, one limitation of synthesizing nanoparticles inside parasites is the inconsistent distribution of accumulated hemozoin within the parasites. The Raman signal acquired from a location with parasites and nanoparticles inside would be much stronger than that from another location without them. Because the distribution of parasites with nanoparticles inside is discrete and usually inhomogeneous in the sample, the variation in Raman peak intensities from random locations or different batches is higher than that in Method 1. For the same reason, there is no strong correlation between the SERS peak intensity and the parasitemia level. However, with the lowest detectable parasitemia level at 0.00005%, Method 2 can be utilized to determine whether a patient is infection positive or to screen high-risk population.
The simplicity and speed of our SERS based techniques indicate their great promise in malaria diagnosis. In the case of P. falciparum infections in which parasites observed in peripheral blood are predominantly in the ring stage, Method 1 would allow the relatively accurate determination of the parasitemia level. While infections containing asynchronous stages would be more difficult to accurately quantify as the hemozoin level varies between the different stages unless an extra synchronization step is employed.
One possible challenge for SERS based techniques that rely on solely hemozoin measurements is its inability to distinguish between hemozoin produced by a viable parasite and that still circulating in blood due to a prior infection. One earlier work has shown that detection of residual hemozoin using a Cell-Dyn automated haematology analyser from a prior malaria infection can be achieved up to 4 weeks after treatment5. This suggests the more sensitive SERS technique would also detect hemozoin from past malaria infections, which will somewhat limit the ability of this approach to making a definitive diagnosis of an active malaria infection. However the technique can be used to accurately exclude a malaria infection and provide crucial data of either a recent or current asymptomatic infection. This information would be of immense value in the later stages of a malaria eradication campaign43,44.
Compared to the previously published SERS method33, our strategy provides the added benefit of easier nanoparticle preparation, lower cost and higher sensitivity. Moreover, the SERS measurements in our methods were performed on random locations rather than on selected hot spots resulting in increased speed. Although an automated method for hot spot searching has been developed45,46, such a method requires scanning the entire sample and its accuracy can be affected by fluorescence, which makes it time-consuming and inconvenient. The waiver of hot spot search in our techniques would facilitate the development of fast and convenient automated SERS measurements for field use in the future. In addition, Method 1 also provides the possibility to quantify parasitemia levels directly, which was not available in the published SERS method33 possibly due to large variation in SERS peak intensities. While the greater variation of spectral intensities in Method 2 is one limitation, two potential strategies could be used to minimize it. One strategy is to combine Method 2 with an automated hot-spot searching method. With a proper threshold setting, one can scan the entire sample to measure only “hot spots” in the sample to minimize variation. The other potential way is to concentrate the parasites with nanoparticles using an isolating device such as MidiMACS (Miltennyi Biotec, Bergisch Gladbach, Germany), to minimize the chance of measuring regions that contain no parasites or nanoparticles.
The improvements described here now make SERS comparable to the standard microscopy method in terms of detection limit but without the need of an experienced operator. Most steps in the two methods reported in this study could be implemented in a microfluidic chip to enable automatic sample preparation. A paper based microfluidic chip38 could further lower the cost of sample preparation. The bottleneck in the high cost of a Raman spectrometer with decent spectral resolution could be overcome by a much cheaper alternative method, in which a cost effective and compact Raman spectrometer37 acquires raw Raman spectra with low spectral resolution and a spectral recovery method we developed recently47 is used to reconstruct the Raman spectra with high spectral resolution. Therefore the two SERS based methods developed in this study in combination with the cost effective Raman spectrometer we are currently developing hold great potential for malaria diagnosis even in low-resource regions.
Methodology
Parasite culture
Plasmodium falciparum parasite (3D7) were cultured in fresh red blood cells (RBCs) at 5% hematocrit with media constituted of bicarbonate buffered RPMI 1640 (RPMI 1640 Medium, Life Technologies, Grand Island, USA) supplemented with 5% albumax (AlbuMAX® I Lipid-Rich BSA, Life Technologies, Grand Island, USA), 200 μM hypoxanthine and 20 μg/ml gentamycin as described previously48. The cells were maintained at 37 °C and a gas mixture of 5% CO2, 1% O2 and 94% N2. After 5 cycles of parasite growth, the mixed culture was synchronized using 5% D-sorbitol (D-Sorbitol, Sigma, St. Louis, USA) treatment49 and allowed to grow one more cycle then were washed to get rid of floating hemozoin. Parasites were mounted onto glass slides and stained with Giemsa (Giemsa stain, Sigma, St. Louis, USA). The parasitemia level was estimated by manual counting under microscopic observation after Giemsa staining and confirmed by staining cells with syber-green (SYBER®Green, Life Technologies, Grand Island, USA) dye and counting with BD LSRII FACS (BD Biosciences, San Jose, USA). Appropriate dilutions were made using non-infected blood in RPMI 140 for further analysis. Optical microscopy was used to confirm the stages of parasites. It turned out that nearly 98% of parasites were in the ring stage and 2% in the early trophozoite stage in this study.
Sample preparation in Method 1 (synthesizing nanoparticles separately)
In Method 1, silver nanoparticles were synthesized separately and then mixed with isolated hemozoin. Silver nanoparticles were synthesized using the reduction method31,34,50. A total of 33-μl Triton X-100 (Triton X-100 Detergent, Bio-Rad Laboratories, Hercules, USA) was mixed with 5-ml hydroxylamine hydrochloride (0.03 mM, MP Biomedicals, Santa Ana, USA) and NaOH (0.15 mM). The mixture was added drop-wise over a period of 8 min to 45 ml aqueous AgNO3 (Silver nitrate, Merck, Kenilworth, USA) with a concentration of 1.11 mM. The resulting solution was then sonicated (Elma E30H, Elma, Wetzikon, Switzerland) for 30 min. Finally, the generated silver nanoparticles were centrifuged and resuspended in 5 ml solution. The suspension was sonicated for 5 min for later use.
Hemozoin crystals were extracted by simple cell lysis and centrifuging. A total of 10 μl infected blood sample were dispersed in 50 ml deionized water mixed with 100 μl Triton X-100 and then sonicated for 5 min. Released hemozoin crystals were collected by centrifuging at 5000 rpm for 5 min (Sartorius 2-16, Sigma Laborzentrifugen, Ostrode, Germany) and resuspended in 5-ml NaOH solution (0.05 mM).
Then silver nanoparticles suspension and hemozoin suspension were mixed together (1:1 v/v). The mixture was sonicated for another 2 min. During Raman measurements, the sample was smeared on a slide covered by aluminum foil as the substrate. On each slide, five random locations were selected for Raman measurements while the sample was still wet.
Sample preparation in Method 2 (synthesizing nanoparticles inside parasites)
In Method 2, silver nanoparticles were synthesized inside parasites directly to achieve closer contact with hemozoin than Method 1. It should be highlighted that this method keeps the local hemozoin concentration in parasites because it does not break down parasites. A total of 10-μl lysed blood was dispersed in 50-ml deionized water and then sonicated for 5 min to achieve blood cell lysis. Different from the cell lysis procedure in Method 1, Triton X-100 was removed in the lysis step of Method 2 to prevent the lysis of parasites. The mixture was pushed through a filter with a pore size of 0.2 μm (Supor Syringe filters, Pall Life Science, Washington, USA) in a syringe to remove hemoglobin in the lysed blood sample. Then, the residue in the filter was flushed out and suspended in 45-ml AgNO3 (Silver nitrate, Merck, Kenilworth, USA) solution with a concentration of 1.11 mM. Then 33-μl Triton X-100 (Triton X-100 Detergent, Bio-Rad Laboratories, Hercules, USA) was mixed with 5-ml solution of hydroxylamine hydrochloride (0.03 mM, MP Biomedicals, Santa Ana, USA) and NaOH (0.15 mM), which was then added drop-wise over a period of 8 min to aqueous AgNO3 to reduce silver. In this step, the volume of Triton X-100 was low and it mostly bonded to silver nanoparticles to prevent them from aggregation, which limited its effect on cell lysis to a low level. The resulting solution was then sonicated (Elma E30H, Elma, Wetzikon, Switzerland) for 30 min. Finally, the sonicated solution was pushed through a 0.2-μm filter (Supor Syringe filters, 25 mm, 0.2 μm, Pall Life Science, Washington, USA) in a syringe to filter away small excessive nanoparticles and chemicals and leave analyte on the filter paper. The filter paper was taken out and deposited on a slide covered by aluminum foil for Raman measurements. For each filter paper, five random locations were selected for Raman measurements.
Raman measurements and data processing
A micro-Raman spectrometer system (inVia, Renishaw, Aberdeen, UK) coupled with a microscope (Alpha 300, WITec, Ulm, Germany) in a backscattering geometry was used to measure all Raman spectra. The system was coupled to a Czerny-Turner type spectrograph (f = 250 mm) with a holographic grating (1800 grooves/mm) and a RemCam CCD detector (inVia, Renishaw, Aberdeen, UK), which yields a spectral resolution of 2 cm−1. A 633-nm diode laser (Renishaw, Aberdeen, UK) was used for excitation with a focal spot size about 3 μm on the sample through a microscope objective lens (5X, NA = 0.2, Leica, Wetzlar, Germany). The exposure time was 10 seconds and the excitation was 2.5 mW for all SERS measurements. The combination of similar exposure time and excitation power was also used previously in our magnetic enriched SERS study31, which can achieve the tradeoff between the maximization of SERS intensity and minimization of the thermal effect.
In each sample, the Raman spectra were measured from five random locations then averaged to reduce variation. No hot spot search was done during spectral measurements in Method 1 or Method 2. The measurement at each parasitemia level was repeated in seven different samples in Method 1 and five different samples in Method 2 for every concentration to calculate the standard deviation. In each raw spectrum, a fifth-order polynomial was used to fit the fluorescence background and then the polynomial was subtracted from the raw spectrum to generate the clean Raman spectrum.
To obtain the contribution of hemozoin alone in the measurement of an infected blood sample for convenient visualization in Method 1, the clean Raman spectrum of an infected blood sample was modeled as the summation of the contribution from the normal blood sample and that from hemozoin. The Raman spectra of the latter two basic components were measured separately (see Fig. 1 in Supplementary Information), in which the basic component spectrum of hemozoin was replaced by that measured from β-hematin at a concentration 10 μg/ml. This is justified by the fact that β-hematin is equivalent to hemozoin in terms of Raman features29,41 and much easier to synthesize. Then the contribution of each component to the infected blood spectrum was estimated using a least square regression method51 implemented by the lsqcurvefit function in MATLAB with an option of trust-region-reflective algorithm.
Detection limit estimation
To determine the detection limit for Method 1, a t-test was conducted to evaluate the difference in hemozoin contribution between every infected blood sample with a progressively decreasing parasitemia level and the normal blood sample. When there was no difference between the two samples, the parasitemia level in the previous infected blood sample that still demonstrated a significant difference in hemozoin contribution compared to the normal blood sample was determined to be the smallest detectable parasitemia level. The lowest detectable parasitemia level for Method 2 was determined in the same manner by conducting a series of t-tests except that the Raman peak intensity at 1623 cm−1 instead of hemozoin contribution was compared.
Ethics statement
The use of human blood strictly followed protocols and guidelines that were approved by the domain-specific review board of Nanyang Technological University (IRB number: NTU-IRB 11/12/2011). Blood component collection service was provided by Blood Transfusion Service and Blood Donation Centre of National University Hospital. All individuals gave informed consent.
Additional Information
How to cite this article: Chen, K. et al. Towards ultrasensitive malaria diagnosis using surface enhanced Raman spectroscopy. Sci. Rep. 6, 20177; doi: 10.1038/srep20177 (2016).
Change history
23 March 2017
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
WHO. World malaria report 2014. (World Health Organization, 2014).
Weissbuch, I. & Leiserowitz, L. Interplay between malaria, crystalline hemozoin formation and antimalarial drug action and design. Chemical reviews 108, 4899–4914 (2008).
Hänscheid, T., Egan, T. J. & Grobusch, M. P. Haemozoin: from melatonin pigment to drug target, diagnostic tool and immune modulator. The Lancet Infectious Diseases 7, 675–685 (2007).
Wongsrichanalai, C., Barcus, M. J., Muth, S., Sutamihardja, A. & Wernsdorfer, W. H. A review of malaria diagnostic tools: microscopy and rapid diagnostic test (RDT). The American journal of tropical medicine and hygiene 77, 119–127 (2007).
De Langen, A. J. et al. Automated detection of malaria pigment: feasibility for malaria diagnosing in an area with seasonal malaria in northern Namibia. Tropical Medicine & International Health 11, 809–816 (2006).
Newman, D. M. et al. A magneto-optic route toward the in vivo diagnosis of malaria: preliminary results and preclinical trial data. Biophysical journal 95, 994–1000 (2008).
Demirev, P. A. et al. Detection of malaria parasites in blood by laser desorption mass spectrometry. Analytical chemistry 74, 3262–3266 (2002).
Press, G. A., Amaral, D. G. & Squire, L. R. Hippocampal abnormalities in amnesic patients revealed by high-resolution magnetic resonance imaging. Nature 341, 54–57 (1989).
Bruña-Romero, O. et al. Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. International journal for parasitology 31, 1499–1502 (2001).
Perandin, F. et al. Development of a real-time PCR assay for detection of Plasmodium falciparum, Plasmodium vivax and Plasmodium ovale for routine clinical diagnosis. Journal of clinical microbiology 42, 1214–1219 (2004).
Adeoye, G. & Nga, I. Comparison of Quantitative Buffy Coat technique (QBC) with Giemsa-stained Thick Film (GTF) for diagnosis of malaria. Parasitology international 56, 308–312 (2007).
Grimberg, B. T. Methodology and application of flow cytometry for investigation of human malaria parasites. Journal of Immunological Methods 367, 1–16 (2011).
Früh, K., Müller, H.-M., Bujard, H. & Crisanti, A. A new tool for the serodiagnosis of acute Plasmodium falciparum malaria in individuals with primary infection. Journal of immunological methods 122, 25–32 (1989).
Mohanty, S. K., Uppal, A. & Gupta, P. K. Self-rotation of red blood cells in optical tweezers: prospects for high throughput malaria diagnosis. Biotechnology letters 26, 971–974 (2004).
Khoshmanesh, A. et al. Detection and Quantification of Early-Stage Malaria Parasites in Laboratory Infected Erythrocytes by Attenuated Total Reflectance Infrared Spectroscopy and Multivariate Analysis. Analytical Chemistry 86, 4379–4386, 10.1021/ac500199x (2014).
Newman, D. M., Matelon, R. J., Wears, M. L. & Savage, L. B. The in vivo diagnosis of malaria: feasibility study into a magneto-optic fingertip probe. Selected Topics in Quantum Electronics, IEEE Journal of 16, 573–580 (2010).
Wood, B. R. et al. Raman imaging of hemozoin within the food vacuole of Plasmodium falciparum trophozoites. FEBS letters 554, 247–252 (2003).
Cordoliani, Y. S. et al. MR of cerebral malaria. American journal of neuroradiology 19, 871–874 (1998).
Peng, W. K. et al. Micromagnetic resonance relaxometry for rapid label-free malaria diagnosis. Nature medicine 20, 1069–1073 (2014).
Kimura, M. et al. Identification of the four species of human malaria parasites by nested PCR that targets variant sequences in the small subunit rRNA gene. Parasitology International 46, 91–95 (1997).
Hsiang, M. S., Greenhouse, B. & Rosenthal, P. J. Point of Care Testing for Malaria Using LAMP, Loop Mediated Isothermal Amplification. Journal of Infectious Diseases 210, 1167–1169 (2014).
Wood, B. R. et al. Resonance Raman spectroscopy reveals new insight into the electronic structure of β-hematin and malaria pigment. Journal of the American Chemical Society 126, 9233–9239 (2004).
Hobro, A. J., Konishi, A., Coban, C. & Smith, N. I. Raman spectroscopic analysis of malaria disease progression via blood and plasma samples. Analyst 138, 3927–3933 (2013).
Huang, H. et al. Confocal Raman spectroscopic analysis of the cytotoxic response to cisplatin in nasopharyngeal carcinoma cells. Analytical Methods 5, 260–266 (2013).
Dasgupta, R., Shanker Verma, R., Ahlawat, S., Uppal, A. & Kumar Gupta, P. Studies on erythrocytes in malaria infected blood sample with Raman optical tweezers. Journal of Biomedical Optics 16, 077009, 10.1117/1.3600011 (2011).
Frosch, T., Koncarevic, S., Becker, K. & Popp, J. Morphology-sensitive Raman modes of the malaria pigment hemozoin. Analyst 134, 1126–1132 (2009).
Delahunt, C., Horning, M. P., Wilson, B. K., Proctor, J. L. & Hegg, M. C. Limitations of haemozoin-based diagnosis of Plasmodium falciparum using dark-field microscopy. Malaria journal 13, 147 (2014).
Jamjoom, G. A. Dark-field microscopy for detection of malaria in unstained blood films. Journal of clinical microbiology 17, 717–721 (1983).
Pagola, S., Stephens, P. W., Bohle, D. S., Kosar, A. D. & Madsen, S. K. The structure of malaria pigment β-haematin. Nature 404, 307–310 (2000).
Wood, B. R. et al. Tip-enhanced Raman scattering (TERS) from hemozoin crystals within a sectioned erythrocyte. Nano letters 11, 1868–1873 (2011).
Yuen, C. & Liu, Q. Magnetic field enriched surface enhanced resonance Raman spectroscopy for early malaria diagnosis. Journal of Biomedical Optics 17, 0170051–0170057, 10.1117/1.JBO.17.1.017005 (2012).
Bonifacio, A. et al. Spatial distribution of heme species in erythrocytes infected with Plasmodium falciparum by use of resonance Raman imaging and multivariate analysis. Analytical and bioanalytical chemistry 392, 1277–1282 (2008).
Garrett, N. L. et al. Bio-sensing with butterfly wings: naturally occurring nano-structures for SERS-based malaria parasite detection. Physical Chemistry Chemical Physics. http://dx.doi.org/10.1039/C4CP04930F (2015).
Yuen, C. & Liu, Q. Optimization of Fe3O4@Ag nanoshells in magnetic field-enriched surface-enhanced resonance Raman scattering for malaria diagnosis. Analyst 138, 6494–6500 (2013).
Charan, S. et al. Synthesis of surface enhanced Raman scattering active magnetic nanoparticles for cell labeling and sorting. Journal of Applied Physics 105, 07B310 (2009).
Zhai, Y. et al. Fabrication of iron oxide core/gold shell submicrometer spheres with nanoscale surface roughness for efficient surface-enhanced Raman scattering. The Journal of Physical Chemistry C 113, 7009–7014 (2009).
Malinen, J. et al. Advances in miniature spectrometer and sensor development. Next-Generation Spectroscopic Technologies VII; Proc. SPIE 9101; 2014. p. 91010C.
Wu, Y., Xue, P., Hui, K. M. & Kang, Y. A paper-based microfluidic electrochemical immunodevice integrated with amplification-by-polymerization for the ultrasensitive multiplexed detection of cancer biomarkers. Biosensors and Bioelectronics 52, 180–187 (2014).
Alayash, A. I., Patel, R. P. & Cashon, R. E. Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxidants and Redox Signaling 3, 313–327 (2001).
Luchsinger, B. P. et al. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the β subunits. Proceedings of the National Academy of Sciences 100, 461–466 (2003).
Frosch, T. et al. In situ localization and structural analysis of the malaria pigment hemozoin. The Journal of Physical Chemistry B 111, 11047–11056 (2007).
Price, R. N. et al. Global extent of chloroquine-resistant Plasmodium vivax: a systematic review and meta-analysis. The Lancet Infectious Diseases 14, 982–991, http://dx.doi.org/10.1016/S1473-3099(14)70855-2 (2014).
Mendis, K. et al. From malaria control to eradication: The WHO perspective. Tropical Medicine & International Health 14, 802–809 (2009).
Alonso, P. L. et al. A research agenda to underpin malaria eradication. PLoS medicine 8, e1000406 (2011).
Pavillon, N., Bando, K., Fujita, K. & Smith, N. I. Feature-based recognition of Surface-enhanced Raman spectra for biological targets. J. Biophotonics 6, 587–597, 10.1002/jbio.201200181 (2013).
Smith, N. I. et al. Laser-targeted photofabrication of gold nanoparticles inside cells. Nat Commun 5, 10.1038/ncomms6144 (2014).
Chen, S. et al. Recovery of Raman spectra with low signal-to-noise ratio using Wiener estimation. Optics Express 22, 12102–12114 (2014).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
Aley, S., Sherwood, J., Marsh, K., Eidelman, O. & Howard, R. Identification of isolate-specific proteins on sorbitol-enriched Plasmodium falciparum infected erythrocytes from Gambian patients. Parasitology 92, 511–525 (1986).
Pastoriza-Santos, I. & Liz-Marzán, L. M. Formation and stabilization of silver nanoparticles through reduction by N, N-dimethylformamide. Langmuir 15, 948–951 (1999).
Ong, Y. H., Lim, M. & Liu, Q. Comparison of principal component analysis and biochemical component analysis in Raman spectroscopy for the discrimination of apoptosis and necrosis in K562 leukemia cells. Optics express 20, 22158–22171 (2012).
Moody, A. Rapid diagnostic tests for malaria parasites. Clinical microbiology reviews 15, 66–78 (2002).
Gligorijevic, B., McAllister, R., Urbach, J. S. & Roepe, P. D. Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 1. Quantification of hemozoin development for drug sensitive versus resistant malaria. Biochemistry 45, 12400–12410 (2006).
Sullivan, D. J. Hemozoin: a Biocrystal Synthesized during the Degradation of Hemoglobin. Biopolymers Online 9, 129–163, http://dx.doi.org/10.1002/3527600035.bpol9007 (2005).
Acknowledgements
The authors would like to acknowledge financial support from the following funding sources. C. Y. was supported by Lee Kuan Yew Postdoctoral Fellowship under the Lee Kuan Yew Endowment Fund in Singapore; P. P was supported by Singapore Ministry of Health’s National Medical Research Council under its Individual Research Grant (NMRC/1292/2011); Q. L. was supported by Tier 1 grant (RG38/14) funded by the Ministry of Education in Singapore.
Author information
Authors and Affiliations
Contributions
K.C., C.Y. and Q.L. conceived and designed the study. K.C. performed the experiments. Y.A. provided malaria parasites and took microscopic images. K.C. and Q.L. wrote the paper. K.C., Q.L. and P.P. reviewed and edited the manuscript. All authors approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, K., Yuen, C., Aniweh, Y. et al. Towards ultrasensitive malaria diagnosis using surface enhanced Raman spectroscopy. Sci Rep 6, 20177 (2016). https://doi.org/10.1038/srep20177
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20177
This article is cited by
-
Rapid phenotyping towards personalized malaria medicine
Malaria Journal (2020)
-
Detection of malaria parasites in dried human blood spots using mid-infrared spectroscopy and logistic regression analysis
Malaria Journal (2019)
-
Selecting better diagnostic kits for diagnosis of malarial parasites at point of care
3 Biotech (2019)
-
Sustained and Cost Effective Silver Substrate for Surface Enhanced Raman Spectroscopy Based Biosensing
Scientific Reports (2017)
-
“Optical and Surface Enhanced Raman Scattering properties of Ag modified silicon double nanocone array”
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
