
Abstract
Using transcriptome data to mine microsatellite and develop markers has growingly become prevalent. However, characterizing the possible function of microsatellite is relatively rare. In this study, we explored microsatellites in the transcriptome of the brown alga Sargassum thunbergii and characterized the frequencies, distribution, function and evolution and developed primers to validate these microsatellites. Our results showed that Tri-nucleotide is the most abundant, followed by di- and mono-nucleotide. The length of microsatellite was significantly affected by the repeat motif size. The density of microsatellite in the CDS region is significantly lower than that in the UTR region. The annotation of the transcripts containing microsatellite showed that 573 transcripts have GO terms and can be categorized into 42 groups. Pathways enrichment showed that microsatellites were significantly overrepresented in the genes involved in pathways such as Ubiquitin mediated proteolysis, RNA degradation, Spliceosome, etc. Primers flanking 961 microsatellite loci were designed and among the 30 pairs of primer selected randomly for availability test, 23 were proved to be efficient. These findings provided new insight into the function and evolution of microsatellite in transcriptome and the identified microsatellite loci within the annotated gene will be useful for developing functional markers in S. thunbergii.
Similar content being viewed by others


Introduction
Microsatellites, also known as simple sequence repeats (SSR) or short tandem repeats (STR), are repeated DNA sequences comprising tandem array of short motifs (generally, 1–6 nucleotides), which are widely spread in both eukaryotic and prokaryotic genomes1,2. Microsatellite was initially used as robust molecular marker and received intensive attention for geneticist and evolutionary biologist. Microsatellite marker is codominant, abundant, multi-allelic and can be detected by simple and reproducible assays3. These important attributes have enabled microsatellite to be powerful marker for molecular population genetics, marker-assisted breeding, DNA fingerprinting, conservation genetics or QTL mapping and positional cloning of target genes4,5. Despite the wide utilization in the above fields, microsatellite is commonly regarded as “junk” in the genome (i.e. with no significant role as genomic information)6 and relative less studies concentrated on its origin, distribution and evolution in genome1,2,7. Recently, as microsatellites have been increasingly identified and characterized within protein-coding genes and their untranslated regions (UTRs), more and more evidences indicate that mi®crosatellite could play a positive role in adaptive evolution via the molecular and phenotypic effects of microsatellite repeat-number variation6,8.
The main limitation of microsatellite markers development is the requirement of prior characterization of sequences containing microsatellite loci to allow primers design for PCR, making the development of microsatellite markers to be an experimentally long, labor intensive and costly process9. The process involved the construction of genomic libraries using recombinant DNA enriched for a few targeted SSR motifs and the isolation and sequencing of clones containing microsatellite loci10,11. With the advent of next-generation sequencing technologies (NGS), it becomes methodologically efficient and economical to sequence the entire genomes or transcriptomes in greater depth than ever before12,13. Several recent studies have demonstrated that the easiest way to identify microsatellite loci is to mine the genome or transcriptome produced by NGS9. The mechanism of microsatellite evolution and the genome-wide distribution were not well understood yet in plants mostly due to the lack of genomic information. The microsatellite identification and characterization in large-scale in genome or transcriptome can thus provide important resource and opportunity to study these themes, together with the development of new genetic markers. In addition, repeat-number variation of microsatellite loci located in or linkage to function genes have impact on chromatin organization, regulation of gene activity, recombination, DNA replication, cell cycle, mismatch repair system1,7. Previous studies suggest that microsatellites from transcriptome may facilitate us to evaluate the association between microsatellite marker and functional genes or phenotypes1.
Sargassum thunbergii is an ecologically and economically important brown macroalga. It widely spreads in the intertidal and shallow sublittoral zone along the coasts of China, Japan and Korea14. This species usually grows luxuriantly and can form seaweed forest together with other Sargassum spp. and kelp, acting as spawning, nursery and feeding ground for marine animals15,16. It is good raw material as well for alginate, mannitol, polyphenol and other bioactive substances widely used in chemical, pharmaceutical and food products17,18,19. More recently, S. thunbergii has been proposed as a promising candidate alga to construct macroalgal bed in the intertidal zone because of its high tolerance to thermal, osmotic and desiccation stress. In China, the wild resources of S. thunbergii were largely eradicated with the rapid growth of holothurian aquaculture industry, as it was regarded as the best natural feed for holothurian. Over the last decade, the technologies for artificial seedling rearing and commercial cultivation of S. thunbergii have been developed in China20,21.
The ecological and economical value of S. thunbergii boosted a growing number of studies, especially in the terms of population genetics and genetic improvement, have been conducted to support the development of cultivation industry22. To accelerate gene discovery and elucidate the molecular mechanism of special biological processes and ecophysiological characteristics in S. thunbergii, we recently sequenced and assembled de novo the transcriptome23. In the present study, we used the transcriptome data to mine microsatellite loci and characterize their frequency, distribution and function and then, we design primers for the amplifying of microsatellite loci and validate the availability of some randomly selected primers. The findings herein will help us to better understand microsatellite evolution in S. thunbergii transcriptome and the developed microsatellite markers can meet the urgent need for studies of population genetics, genetic mapping and functional gene cloning in Sargassum species.
Results
Overall characteristics of microsatellite in S. thunbergii transcriptome
A total of 46,269 expression sequence tags, obtained from the de novo transcriptome sequencing of S. thunbergii, were assembled further after redundancy elimination and produced 36,119 consensus sequences with average length of 1,196 bp and N50 of 1,851 bp. These consensus sequences were divided into two groups: 26,451 singletons and 4087 clusters (with over 70% similarities among sequences in clusters) consisting of 9,668 sequences. As showed in the Table 1, a total of 2915 microsatellite loci were identified. Among the 36,119 sequences examined, 2528 (6.70%) harbored microsatellite locus, while only 322 (0.89%) contain more than one microsatellite locus. Most (2822, 96.8%) of the microsatellite loci are the pure or perfect ones, while a small proportion (93, 3.19%) were the compound microsatellites. The frequency or density of microsatellite in S. thunbergii transcriptome was 0.068 loci per Kbp.
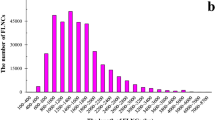
Out of the total 2915 SSRs, 1680, 772 and 276 are tri-, di- and mono-nucleotide repeat motif and 106, 45 and 36 are penta-, tetra- and hexa-nucleotide repeat, respectively. Tri-, di- and mono-nucleotide repeat listed as top three repeat motifs with the largest number, followed by penta-, tetra and hexa-nucleotide (Fig. 1). For the mono-nucleotide motif, G/C was the most abundant type and the repeat number of mono-nucleotide can reach up to 23 times. For the di-nucleotide motif, AC/GT was the most abundant type with a total of 338 loci, while there was only 11 CG/GC. The length of di-nucleotide tract can reach up to 24 bp implying it can repeat as many as 12 times. There were ten tri-nucleotide motif types, among which AGC/CTG accounted for about 50.8%, while AAT/ATT only accounted for 1.48%. There were 17, 44 and 25 repeat motif types for tetra-, penta and hexa-nucleotide microsatellite, respectively and the different repeat motif types presented quite evenly.

The number distribution of different microsatellite motif types in Sargassum thunbergii transcriptome.
The average length of the microsatellites was 16.32 bp. The length variation of microsatellite was significantly affected by repeat motif size. Except the length difference between mono- and di-nucleotide as well as tetra- and penta-nucleotide, the length differences among other motif size classes were all statistically significant (P < 0.001). The mono-nucleotide has the shortest average length (13.9 bp), while hexa-nucleotide repeat motif was the longest with an average of 25.9 bp. The longest microsatellite identified was 63 bp, with a tri-nucleotide motif repeated 21 times. In addition, the microsatellite length was not significantly affected by base composition (P > 0.05).
Comparison among microsatellites located in CDS, 5′ UTR and 3′ UTR
We investigated the distributional characteristics of microsatellite in the transcript (CDS, 5′ UTR and 3′ UTR). Out of the total 2915 SSRs, 629, 832 and 739 were located in the CDS, 5′ UTR and 3′ UTR, respectively (Table 2). The remaining 715 SSRs were not ascertained because the transcripts containing them lacked enough information to delimit the CDS region. The density or frequency of SSRs in the CDS region is significant lower than that in the UTR region (χ2 = 28.16, P < 0.01). In other words, the UTR regions harbored more microsatellite compared to the CDS. The motif size classes of microsatellite were significantly affected (χ2 = 396.00, P < 0.01) by the location (CDS, 5′ UTR and 3′ UTR). For the microsatellite located in the CDS, most of them (91.23%) were the tri-nucleotide. Although the tri-nucleotide microsatellites also dominated in the UTR, the proportions of mono- and di-nucleotide in the UTR were much higher than that in the CDS. The length of microsatellite tract differs significantly between the CDS and the UTR (P < 0.01). Compared to the UTR, the microsatellites in the CDS were much shorter; however, the difference in microsatellite length was not statistically significant between 5′ UTR and 3′ UTR (P > 0.01).
Function annotation for genes containing microsatellite
To explore the function of microsatellite, the transcripts containing the SSR was annotated. GO assignment was used to classify the transcripts according to their function. Based on sequence homology, 573 microsatellite-containing transcripts had GO annotations and can be categorized into three functional groups and 42 sub-groups (Fig. 2). For the “biological process” groups, there were 19 subgroups, among which “cellular process”, “metabolic process” and “single-organism process” were the top three sub-groups involved the most genes. For the “cellular component” group, there were 13 subgroups, among which “cell”, “cell part” and “organelle” were the top three sub-groups involved the most genes. Ten sub-groups constitute the “molecular function” group and the “catalytic activity”, “binding” and “structural molecular activity” involved the most genes. GO enrichment analysis showed that five GO terms (GO:0016747, GO:0004633, GO:0004871, GO:0060089, GO:0042578) over-represented significantly (Q-value < 0.05). The genes involved in the five enriched GO terms possessed the following function respectively: transferring acyl group activity, phosphopantothenoylcysteine decarboxylase activity, signal transducer activity, phosphoric ester hydrolase activity. The function of microsatellite-containing transcripts was further surveyed by the KEGG pathway analysis. The results showed that the transcripts involved in 94 pathways totally. After enrichment analysis, four pathways, that is ko04120 (Ubiquitin mediated proteolysis), ko03018 (RNA degradation), ko03040 (Spliceosome) and ko00900 (Terpenoid backbone biosynthesis), were obtained (Fig. 3).

The classification of genes containing microsatellite locus based on the Gene ontology (GO) annotation.

The top 20 enriched pathways involved the gens containing microsatellite locus.
Primers design and validation for microsatellite markers
Based on the transcripts containing microsatellite loci, primers flanking 961 microsatellite loci were successfully designed (Table S3). A total of 30 pairs of primer were randomly selected and used to test the availability of these designed primers in a mixed population comprising six individuals. The results showed that seven pairs of primer gave no amplicon, whereas 23 pairs of primer could amplify successfully. Out of the 23 pairs of primer, 21 produced the amplicon with expected size, while 2 gave amplicons larger than the expected size. The 21 pairs of primer were thus used to assess the genetic diversity of a mixed population which consists of six geographic subpopulations with each having five individual. Ten pairs of primer could amplify polymorphic SSR alleles (Table 3). These microsatellite loci in the tested population possessed diverse number of alleles (2~5) with an average of 3.6 (Table 3). Polymorphic Information Content (PIC) of microsatellite markers ranged from 0.339 to 0.694, suggesting that these SSR markers could be used as robust molecular markers for future population genetics, evolutionary analysis or other applications. It is worthy to mention that only three loci (SW9, SW17 and SW18) were in the Hardy-Weinberg equilibrium in the mixed population. Linkage disequilibrium test showed that most of the loci were in linkage equilibrium except three pairs of loci (SW6 and SW17, SW6 and SW18, SW16 and SW35).
Discussion
In this study, microsatellites in the transcriptome of S. thunbergii were mined and characterized. The results showed that microsatellites were only presented in a small proportion of the transcripts (6.70%), consisting with the estimation that 2–11% transcripts contain microsatellite24. Recently, more and more ESTs or transcriptome assembled de novo were used to mine microsatellite, including several seaweeds such as Saccharina japonica25,26, Laminaria digitata27, Pyropia28, etc. Although the frequency or density of microsatellite differs slightly in different species due to the varied criteria used in microsatellite identification, it demonstrated that the transcriptome is an invaluable resource for microsatellite identification. These identified microsatellite loci can promote the molecular maker development in S. thunbergii and have potential application in other Sargassaceae species.
Our results showed that microsatellite loci in S. thunbergii transcriptome do not distribute evenly in UTR and CDS regions, with more prevalence in the former than in the latter. The relative prevalence of microsatellites in UTRs was consistent with the results of previous transcriptome surveys29,30,31,32. The density of microsatellite in genome may be depended on or affected by two factors: 1) evolutionary constrains on microsatellite due to the harmful effect of microsatellite mutation on gene function and 2) the direction selection on microsatellites with adaptive roles33,34,35. As microsatellites are highly prone to “indel” mutations by means of slip-strand mispairing33, microsatellites in the CDS region are more likely to damage normal gene function than microsatellite in the UTR regions, leading to higher evolutionary constrains on microsatellites in the CDS region. Conversely, the URT regions exhibited higher tolerance for mutation and possessed higher prevalence of microsatellite due to the lower evolutionary constrains. Moreover, microsatellites in the UTRs may have the “tuning ability” on functional genes34,35, conferring the genes adaptive roles in evolution. Thus, the directional selection on these microsatellites probably favored microsatellite expansion in UTRs.
We found that the Tri-nucleotide was the most abundant among the six motif size classes, consistent with the studies in plants such as Arabidopsis thaliana and rice29, Brassica rapa31, Medicago tunculata30 and Helianthus annuus32. The higher prevalence of Tri-nucleotide may be due to that this type of microsatellites should be less likely to cause frameshift mutations36. The length of microsatellite tract also reflects the effect of evolution and selection on microsatellite loci development. Our study found that the length variation of microsatellite tract was significantly affected by both the repeat motif size classes and the location (in CDS or UTR). Compared to other motif size classes, tri- and hexa-nucleotide were much longer, suggesting the lower evolution constrains on tri- and hexa-nucleotide because these two type microsatellite do not cause frameshift mutation in genes. Moreover, microsatellites in UTR were much longer than those in CDS, reflecting higher evolution constrains on the microsatellites in CDS than in UTR regions.
SSRs are previously regarded as ‘junk’ in genome or as evolutionarily neutral DNA markers. However, microsatellites have growingly been found and characterized within protein-coding genes and their untranslated regions, providing multiple lines of evidence for the function and evolution of microsatellite7,8. When microsatellites locate in or link to functional gene, their repeat motif variation, which is frequently and reversibly by adding or subtracting motif, will influence on gene regulation, transcription, translation and protein function6. In this study, we found that microsatellite were over-represented in genes involved in pathways such as Ubiquitin mediated proteolysis, RNA degradation, Spliceosome and Terpenoid backbone biosynthesis (Fig. 3). The ubiquitin proteolytic system plays important roles in a broad array of basic cellular processes by selective proteolysis and in plant response and adaptation to drought, salinity, cold and nutrient deprivation37. S. thunbergii inhabits in the intertidal zone and frequently suffers severe adverse conditions such as thermal, osmotic, illumination and desiccation stresses as tides rise and fall38. Previous study showed that S. thunbergii possesses innate tolerance to these stresses16, allowing us to postulate that microsatellite located in genes related to Ubiquitin mediated proteolysis may be ecologically crucial for S. thunbergii to accumulate adaptive genetic variation to adapt to harsh environment variables. Microsatellite was also proved to be related to stress adaptation in other organisms. For example, the microsatellites identified in Bemisia tabaci transcriptome were located in the genes related to resistance to environmental stresses and insecticides such as aldehyde oxidase, cytochrome P450 and mitogen-stress activated protein kinases39. The significant enrichment of microsatellite in Helianthus annuus transcriptome was observed in GO terms associated with biological processes that involved in plant response to stress, biotic and abiotic stimuli32.
We also found that partial transcripts harboring microsatellite have the transcription factor activity. A similar investigation of microsatellite in the genome of rice and Arabidopsis showed that some transcripts harboring microsatellite were also related to the transcription factors29. In Elaeis guineensis, microsatellite polymorphisms were found in sequences encoding AP2-like, bZIP, zinc finger, MADS-box and NAC-like transcription factors40. Transcription factors, as the crucial factors in transcription regulation, play a crucial role in plant growth, development and evolution41. Microsatellite in transcription factor coding gene may function as important “Tuning Knob” in evolution34,35. Moreover, microsatellites were also overrepresented in other pathways (Fig. 3), such as “RNA transport”, “Ribosome biogenesis in eukaryotes”, “regulation of autophagy”, although they were not significantly enriched (Q-value > 0.05). In brief, the microsatellite loci and their association to gene function or pathways may shed light on the function of microsatellite. However, because of the limitation of the hypergeometric test used here, it must be careful to make a final conclusion about the function of microsatellite loci. It needs more genetic experiments to validate the probable function annotated by bioinformatics method.
Numerous studies have demonstrated that the EST or transcriptome produced by NGS was valuable resources to efficiently develop SSR markers in large-scale9. Herein, we identified applicable microsatellite loci and some of them have been proved as efficient molecular markers. To the best of our knowledge, these are the first set of microsatellites identified in S. thunbergii. Given the high transferability of genic- or EST-SSR markers, the microsatellites identified from S. thunbergii will have wide application in other Sargassum species with limited number of SSR markers42,43. Previous study revealed low to moderate levels of genetic variations (HE: average expected heterozygosity, varying from 0.2729 ~ 0.2903) within S. thunbergii population using random amplified polymorphic DNA (RAPD) and inter-simple sequence repeat (ISSR) markers22. With the ten microsatellite markers, we found the genetic diversity of S. thunbergii population was much higher (HE: 0.422-0.754). Microsatellite markers can generally reveal higher genetic diversity than RAPD markers3. However, one noteworthy factor here is that the tested population was a mixed population consisting of six geographic subpopulations (from Liaoning, Shandong and Zhejiang Province, China) with each having five individuals, whereas the populations in the previous study were the local populations in Shandong Province22.
Materials and Methods
Microsatellite identification and characterization
A total of 46,269 expression sequence tags, obtained from the de novo transcriptome sequencing of Sargassum thunbergii23, were further assembled with Phrap (http://www.phrap.org/) after redundancy elimination by TIGR v2.144. These assembled consensus sequences were used to identify microsatellite loci using software MISA (Microsatellite searching tool, http://pgrc.ipk-gatersleben.de/misa/) with the following criteria: mono-nucleotide repeats motif with at least 12 repeats, di-nucleotide with six, tri- and quad-nucleotide with five, penta and hexa-nucleotide with four. The criterion for compound microsatellites is that the interval between two repeat motifs was shorter than 100 nt.
In order to investigate the distribution of microsatellite in S. thunbergii transcriptome, the relative position of microsatellites with regard to start and stop codons was inferred to determine whether microsatellites were in 5′UTR, CDS or 3′UTR. The position of start and stop codon in sequences were inferred by comparing potentially homologous sequences in Swissprot database, using the software package ESTScan ver. 2.045,46. The location of microsatellite was determined based on the predicted CDS, 5′ UTR and 3′ UTR region. In order to understand the evolution of microsatellites in S. thunbergii transcriptome, several characteristics of microsatellite, such as, prevalence or density (one loci per Kbp), motif size (motif length) , motif type (base composition), were determined and compared with each other among microsatellite loci located in CDS, 5′ UTR and 3′ UTR. Chi-square analyses were conducted to test whether the density of microsatellite in different transcript domains (CDS, 5′ UTR and 3′ UTR) is significant according to the previously reported method32. Kruskal–Wallis rank sum test47 was conducted to test whether the microsatellite length was affected by (i) transcript domains, (ii) motif size and (iii) motif type. The detailed analysis method and process followed the previous report32.
Function annotation of genes containing SSR
To understand the possible function of microsatellite, all the transcripts harboring microsatellite were searched against the GenBank nr protein database using BLASTx with an E-value cut-off of 10−5. Blast2GO program48 was used to get GO annotation and WEGO software49 to classify the transcripts. To investigate whether some GOs or pathways exhibited microsatellite enrichment, hypergeometric tests were used to determine whether microsatellite-containing transcripts ascribed to specific GOs or pathways are more likely to encode microsatellites than expected by chance. The calculating formula for the P-value is as follows:

where N is the number of all transcripts that with GO or KEGG annotation, n is the number of transcripts containing microsatellite in N, M is the number of all genes annotated to specific GO or pathways and m is number of transcripts containing microsatellite in M. Q-value was obtained from P-value by Bonferroni correction. GOEAST50 was used to carry out GO enrichment analysis, identifying the overrepresented GO terms. For pathway enrichment analysis, all transcripts harboring microsatellite were assigned to terms in KEGG database51 and searched for significantly enriched KEGG terms compared to the whole transcriptome background. The statistical analysis was accomplished by a customized Perl script with the same formula mentioned above.
Primer design and validation for microsatellite loci
To explore the diversity and mutability of the microsatellite loci identified in the S. thunbergii transcriptome, the primers were designed based on the sequences flanking the microsatellite loci using Primer3-2.3.4 with default parameters. A total of 30 pairs of primer were selected randomly and used to amplify the microsatellite loci in a mixed population comprising six geographic subpopulations with each having five individuals (Table S1). Genomic DNA was extracted using a Pant genomic DNA kit (Tiangen Biotech CO., Ltd, Beijing, China) according to the manufacturer’s instructions. Polymerase chain reactions (PCRs) were carried out in a total volume of 20 μL containing 0.5 U Taq DNA polymerase (MBI), 1 × PCR buffer, 0.2 mM dNTP mix, 0.5 μM of each primer set, 2.0 mM MgCl2 and about 50 ng template DNA. The mixture was subjected to 94 °C for 4 min, following by 35 cycles of 30 min at 94 °C, 30 s at annealing temperatures (refer to Table 2), 40 s min at 72 °C and a final step at 72 °C for 10 min. PCR products were resolved via 6% denaturing polyacrylamide gel and visualized by silver-staining52. The observed number of alleles (Na), the mean observed heterozygosity (HO) and the mean expected heterozygosity (HE) for each locus in the tested population were calculated by genetic analysis package POPGENE version 1.353. Tests of the Hardy-Weinberg equilibrium and linkage disequilibrium for these loci in the test population were performed using GENEPOP54.
Additional Information
How to cite this article: Liu, F. et al. Distribution, function and evolution characterization of microsatellite in Sargassum thunbergii (Fucales, Phaeophyta) transcriptome and their application in marker development. Sci. Rep. 6, 18947; doi: 10.1038/srep18947 (2016).
References
Li, Y. C., Korol, A. B., Fahima, T., Beiles, A. & Nevo, E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 11, 2453–2465 (2002).
Oliveira, E. J., Pádua, J. G., Zucchi, M. I., Vencovsky, R. & Vieira, M. L. C. Origin, evolution and genome distribution of microsatellites. Genet. Molecul. Biol. 29, 294–307 (2006).
Powell, W., Machray, G. & Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1, 215–222 (1996).
Tautz, D. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucl. Acid. Res. 17, 6463–6471 (1989).
Miah, G. et al. A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance. Int. J. Mol. Sci. 14, 22499–528 (2013).
Kashi, Y. & King, D. Simple sequence repeats as advantageous mutatorsin evolution. Trends Genet. 22, 253–259 (2006).
Li, Y. C., Korol, A. B., Fahima, T. & Neno, E. Microsatellites within genes: structure, function and evolution. Mol. Biol. Evol. 21, 991–1007 (2004).
Hancock, J. M. & Simon, M. Simple sequence repeats in proteins and their significance for network evolution. Gene 345, 113–118 (2005).
Zalapa, J. E. et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Amer. J. Bot. 99, 193–208 (2012).
Zane, L., Bargelloni, L. & Patarnello, T. Strategies for microsatellite isolation: a review. Mol. Ecol. 11, 1–16 (2002).
Squirrell, J. et al. How much effort is required to isolate nuclear microsatellites from plants? Mol. Ecol. 12, 1339–1348 (2003).
Varshney, R. K., Nayak, S. N., May, G. D. & Jackson, S. A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotech. 27, 522–530 (2009).
Shendure, J. & Aiden, E. L. The expanding scope of DNA. Nat. biotechnol. 30, 1084–1094 (2012).
Tseng, C. K. Common seaweeds of China. Science Press, Beijing (1983).
Tsukidate, J. Studies on the regenerative ability of the brown algae, Sargassum muticum (Yendo) Fensholt and Sargassum tortile C. Agardh. Hydrobiologia, 116/117, 393–397 (1984).
Chu, S. H. et al. Tolerance of Sargassum thunbergii germlings to thermal, osmotic and desiccation stress. Aquat. Bot. 96, 1–6 (2012).
Itoh, H., Noda, H., Amano, H. & Ito, H. Immunological analysis of inhibition of lung metastases by fucoidan (GIV-A) prepared from brown seaweed Sargassum thunbergii. Anticancer Res. 15, 1937–1947 (1995).
Seo, Y., Park, K. E. & Nam,T. J. Isolation of a New Chromene from the Brown Alga Sargassum thunbergii. B. Kor. Chem. Soc. 28, 1831–1833 (2007).
Yabur, R., Bashan, Y. & Hernandez-Carmona, G. Alginate from the macroalgae Sargassum sinicola as a novel source for microbial immobilization material in wastewater treatment and plant growth promotion. J. Appl. Phycol. 19, 43–53 (2007).
Liu, F. L., Sun, X. T., Wang, W. J., Liang, Z. R. & Wang, F. J. Development of a female-specific RAPD marker for Sargassum thunbergii gender identification using bulked segregant analysis. Aquat. Bot. 102, 79–81 (2012).
Liang, Z. R., Sun, X. T., Wang, F. J., Wang, W. J. & Liu, F. L. Reproductive Biology of Sargassum thunbergii (Fucales, Phaeophyceae). American Journal of Plant Sciences 5, 2574–2581 (2014).
Zhao, F. J., Wang, X. L., Liu, J. D. & Duan, D. L. Population genetic structure of Sargassum thunbergii (Fucales, Phaeophyta) detected by RAPD and ISSR markers. J. Appl. Phycol. 19, 409–416 (2007).
Liu, F. L., Sun, X. T., Wang, W. J., Liang, Z. R. & Wang, F. J. De novo transcriptome analysis-gained insights into physiological and metabolic characteristics of Sargassum thunbergii (Fucales, Phaeophyceae). J. Appl. Phycol. 26, 1519–1526 (2014).
Cordeiro, G. M., Casu, R., Mcintyre, C. L., Manners, J. M. & Henry, R. J. Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci. 160, 1115–1123 (2001).
Liu, F. L., Wang, X. L., Yao, J. T., Fu, W. D. & Duan, D. L. Development of expressed sequence tag-derived microsatellite markers for Saccharina (Laminaria) japonica. J. Appl. Phycol. 22, 109–111 (2010).
Wang, G. L. et al. Development of EST-SSR primers and their practicability test for Laminaria. Acta Oceanol. Sin. 30, 112–117 (2011).
Liu, F. L., Wang, F. J. & Duan, D. L. EST-SSR markers derived from Laminaria digitata and its transferable application in Saccharina japonica. J. Appl. Phycol. 24, 501–505 (2012).
Xie, C. T., Li, B., Xu, Y., Ji, D. H. & Chen, C. S. Characterization of the global transcriptome for Pyropia haitanensis (Bangiales, Rhodophyta) and development of cSSR markers. BMC Genomics, 14, 107 (2013).
Lawson, M. J. & Zhang, L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 7, R14 (2006).
Mun, J. H. et al. Distribution of microsatellites in the genome of Medicago truncatula: a resource of genetic markers that integrate genetic and physical maps. Genetics, 172, 2541–2555 (2006).
Hong, C. P. et al. Genomic distribution of simple sequence repeats in Brassica rapa. Mol. Cells 23, 349–35 (2007).
Pramod, S., Perkins, A. D. & Welch, M. E. Patterns of microsatellite evolution inferred from the Helianthus annuus (Asteraceae) transcriptome. J. Genet. 93, 431–442 (2014).
Levinson, G. & Gutman, G. A. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 (1987).
King, D. G., Soller, M. & Kashi, Y. Evolutionary tuning knobs. Endeavour, 21, 36–40 (1997).
Trifonov, E. N. Tuning function of tandemly repeating sequences: a molecular device for fast adaptation; In Evolutionary theory and processes: modern horizons papers in honour of Eviatar Nevo (ed. S. P. Wasser ), pp. 115–138. Kluwer Academic Publishers, Massachusetts, USA (2004).
Metzgar, D., Bytof, J. & Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 10, 72–80 (2000).
Stone, S. L. The role of ubiquitin and the 26S proteasome in plant abiotic stress signaling. Frontiers in Plant Science, 5, 135 (2014).
Pearson, G. A. et al. An expressed sequence tag analysis of the intertidal brown seaweeds Fucus serratus (L.) and F. vesiculosus (L.) (Heterokontophyta, Phaeophyceae) in response to abiotic stressors. Mar. Biotechnology 12, 195–213 (2010).
Wang, H. L. et al. Developing conversed microsatellite markers and their implications in evolutionary analysis of the Bemisia tabaci complex. Sci. Rep. 4, 6351 (2014).
Tranbarger, T. J. et al. SSR markers in transcripts of genes linked to posttranscriptional and transcriptional regulatory functions during vegetative and reproductive development of Elaeis guineensis. BMC Plant Biology 12, 1 (2012).
Doebley, J. & Lukens, L. Transcriptional regulators and the evolution of plant form. Plant Cell 10, 1075–1082 (1998).
Bi, Y. H., Yang, X., Sun, Z. & Zhou, Z. G. Development and characterization of 12 polymorphic microsatellite markers in Sargassum vachellianum. Conserv. Genet. Resour. 7, 2013–205 (2015).
Shan, T. F., Pang, S. J., Li, J. & Su, L. Isolation and characterization of eight novel microsatellite loci from the brown alga Sargassum horneri. J. Appl. Phycol. DOI: 10.1007/s10811-014-0521-1 (2015).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Iseli, C., Jongeneel, C. V. & Bucher, P. ESTScan: a program for detecting, evaluating and reconstructing potential coding regions in EST sequences. Proceedings of International Conference on Intelligent Systerms for Molecular Biology, 138, 48 (1999).
Lottaz, C., Iseli, C., Jongeneel, C. V. & Bucher, P. Modeling sequencing errors by combining Hidden Markov models. Bioinformatics 19, 103–112 (2003).
Kruskal, W. H. & Wallis, A. W. Use of ranks in one-criterion analysis of variance. J. Am. Stat. Assoc. 47, 583–621 (1952).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucl. Acid. Res. 36, 3420–3435 (2008).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucl. Acid. Res. 34, W293–297 (2006).
Zheng, Q. & Wang, X. J. GOEAST: a web-based software toolkit for gene ontology enrichment analysis. Nucl. Acid. Res. 36, W358–W363 (2008).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucl. Acid. Res. 36 (Database issue): D480–484 (2008).
Bassam, J. B., Caetano-Anolles, G. & Gresshoff, P. M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 196, 80–83 (1991).
Yeh, F. C., Yang, R. C. & Boyle, T. POPGENE version1.32, Microsoft window-base software for population genetic analysis: a quick user’s guide. University of Alberta, Center for International Forestry Research, Alberta, Canada (1999).
Raymond, M. & Rousset, F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Acknowledgements
This research was funded by the 863 Hi-Tech Research and Development Program of China (2012AA10A413) and National Natural Science foundation of China (NO. 31370264). The authors are grateful to the anonymous reviewers for their constructive comments on this paper.
Author information
Authors and Affiliations
Contributions
F.L. Liu, J.J. Li and W.H. Liu: manuscript preparing; Z.M. Hu, X.T. Sun, W.J. Wang: data analysis; J.J. Li, Z.M. Hu and Z.R. Liang: samples collection; F.L. Liu and F.J. Wang: study design.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, F., Hu, Z., Liu, W. et al. Distribution, function and evolution characterization of microsatellite in Sargassum thunbergii (Fucales, Phaeophyta) transcriptome and their application in marker development. Sci Rep 6, 18947 (2016). https://doi.org/10.1038/srep18947
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18947
This article is cited by
-
An empirical analysis of mtSSRs: could microsatellite distribution patterns explain the evolution of mitogenomes in plants?
Functional & Integrative Genomics (2022)
-
De novo assembly of the seed transcriptome and search for potential EST-SSR markers for an endangered, economically important tree species: Elaeagnus mollis Diels
Journal of Forestry Research (2020)
-
Discovery of novel genic-SSR markers from transcriptome dataset of an important non-human primate, Macaca fascicularis
Scientific Reports (2019)
-
Distribution, Function and Polymorphism Characteristics of Microsatellites in Pyropia yezoensis Transcriptome
Journal of Ocean University of China (2019)
-
Comparative assessment of genetic diversity in Albanian olive (Olea europaea L.) using SSRs from anonymous and transcribed genomic regions
Tree Genetics & Genomes (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
