
Abstract
The subclass Acari (mites and ticks) comprises two super-orders: Acariformes and Parasitiformes. Most species of the Parasitiformes known retained the ancestral pattern of mitochondrial (mt) gene arrangement of arthropods and their mt tRNAs have the typical cloverleaf structure. All of the species of the Acariformes known, however, have rearranged mt genomes and truncated mt tRNAs. We sequenced the mt genomes of two species of Eriophyoidea: Phyllocoptes taishanensis and Epitrimerus sabinae. The mt genomes of P. taishanensis and E. sabinae are 13,475 bp and 13,531 bp, respectively, are circular and contain the 37 genes typical of animals; most mt tRNAs are highly truncated in both mites. On the other hand, these two eriophyoid mites have the least rearranged mt genomes seen in the Acariformes. Comparison between eriophyoid mites and other Aacariformes mites showed that: 1) the most recent common ancestor of Acariformes mites retained the ancestral pattern of mt gene arrangement of arthropods with slight modifications; 2) truncation of tRNAs for cysteine, phenylalanine and histidine occurred once in the most recent common ancestor of Acariformes mites whereas truncation of other tRNAs occurred multiple times; and 3) the placement of eriophyoid mites in the order Trombidiformes needs to be reviewed.
Similar content being viewed by others


Introduction
Eriophyoidea is the largest superfamily in the subclass Acari (mites and ticks), with more than 4,400 described species1. Eriophyoid mites have exceptional morphological and biological characters in comparison to other mites and ticks: two pairs of legs (instead of four pairs), small body size (~200 μm body length) and high host specificity2, to name a few. Some species of eriophyoid mites are pests to crops3 and forests4, such as the wheat curl mite, Aceria tosichella5,6. Other than causing yield loss, some eriophyoid mites can transmit and spread viruses, which can cause further damages to plants6,7.
The typical animal mt genome is circular, 15–20 kb in length with 37 genes8 and was found in most mites and ticks investigated to date except for Steganacarus magnus (16 tRNA genes not identified, possibly due to tRNA truncation)9, Metaseiulus occidentalis (nad3 and nad6 not identified, 18 genes duplicated)10 and Leptotrombidium pallidum (2 genes duplicated)11. Of the 39 species from the superorder Parasitiformes investigated to date, 19 species retained the ancestral pattern of mt gene arrangement for arthropods; these species are from four families: Argasidae (soft ticks, 11 species)12,13,14, Allothyridae (holothyroid mites, 1 species)12, Nuttalliellidae (hard ticks, 1 species)13 and Ixodidae (hard ticks, 6 species)15,16,17. Rearrangement of mt genes was found in the other 20 species from five families: Ixodidae (16 species)18,19, Varroidae (1 species)20, Phytoseiidae (2 species)10,21 and Ologamasidae (1 species)22. In contrast to Parasitiformes, all of the 27 species from the other superorder, Acariformes, have rearranged or highly rearranged mt genomes (details below).
The mt tRNAs of animals usually possess a cloverleaf secondary structure composed of four arms: AA-arm, D-arm, AC-arm and T-arm23. The only exception is the tRNA for serine (anticodon GCT), which lost D-arm in nearly all animals; this is apparently an ancestral feature for animals23. Loss of D-arm or T-arm occurred in other tRNAs but was not common in animals. Large-scale tRNA truncation was first found in nematodes, in which 20 of the 22 tRNAs lack T-arm and the two tRNAs for serine (anticodons GCT and TGA) lack the D-arm24. Later, truncated tRNAs were also found in Acariformes11, Araneae25,26,27, Pseudoscorpiones28, Scorpiones29,30, Thelyphonida30, Acanthocephala31, Insecta32 and Protura33. All of the 25 species of Acariformes mites known have many truncated tRNA genes whereas species from the other superorder, Parasitiformes, do not have truncated tRNAs except for the tRNA for serine (anticodon GCT) and the tRNA for cysteine, which lacks D-arm in Varroa destructor20, Phytoseiulus persimilis21, M. occidentalis21, Haemaphysalis flava14, Rhipicephalus microplus14 and R. sanguineus14.
Prior to this study, no complete mt genomes have been investigated for eriophyoid mites. To understand the evolution of mt genomes in the Acariformes, we sequenced the mt genomes of two eriophyoid mites, Phyllocoptes taishanensis Xue & Hong, 2005 (Eriophyidae: Phyllocoptini) and Epitrimerus sabinae Xue & Hong, 2005 (Eriophyidae: Phyllocoptini). We found rearrangement of mt genes in both eriophyoid mites relative to the hypothetical ancestor of arthropods. Further, both species have highly truncated tRNAs. Here, we present the novel features of the mt genomes of P. taishanensis and E. sabinae and discuss the evolution of mt genomes in the Acariformes, tRNA truncation and the phylogeny of eriophyoid mites in the light of new evidence from these two mites.
Results
General features of the mt genomes of the two eriophyoid mites, P. taishanensis and E. sabinae
The mt genomes of P. taishanensis and E. sabinae are 13, 475 bp and 13, 531 bp long respectively, are circular and have 37 genes: 13 protein-coding genes (PCG), two ribosomal RNA (rRNA) genes and 22 transfer RNA (tRNA) genes (Fig. 1; Table 1). Genes are on both strands: one strand has 27 genes whereas the other strand has 10 genes. The start codons of the 13 PCGs were ATN in E. sabinae. In P. taishanensis, 11 of the 13 PCGs use ATN as start codons whereas cox1 and atp8 appear to start with CTG, which is a rare start codon for animal mitochondria (http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/index.cgi?chapter=cgencodes). We noticed that in the water mites, Unionicola parkeri and U. foili, another rare start codon, TTG, was used34,35. The stop codons were TAA or TAG in both eriophyoid species; incomplete stop codons, T, was found in protein-coding genes that precede a tRNA gene. In both species of eriophyoid mites, TAA was the most common stop codon and was used in nine of the 13 PCGs in P. taishanensis and 10 of the 13 PCGs in E. sabinae (Table 1). The putative control region (CR) of P. taishanensis is short, only 47 bp in size, lying between rrnL and trnW. The putative CR of E. sabinae is 94 bp and is in a different location between trnK and atp6. No conserved regions were found between the CRs of the two eriophyoid mites. No other non-coding regions longer than 16 bp were found in the mt genomes of these two mites.

Map of the mitochondrial genomes of Phyllocoptes taishanensis (A) and Epitrimerus sabinae (B).
Protein-coding genes are color-coded (cox: blue; nad: green; atp: orange; cob: yellow); rRNA genes are in grey; control regions are in black; tRNA genes are in red or purple. Abbreviations of protein-coding genes are: atp6 and atp8 for ATP synthase subunits 6 and 8, cox1–3 for cytochrome oxidase subunits 1–3, cob for cytochrome b, nad1-6 and nad4L for NADH dehydrogenase subunits 1-6 and 4 L, rrnL and rrnS for large and small rRNA subunits. tRNA genes are indicated by the single letter IUPAC-IUB abbreviations for their corresponding amino acids. Arrows and arrowheads show the direction of gene transcription. Numbers at gene junctions indicate the length of non-coding sequences; negative numbers indicate overlap between genes.
Eriophyoid mites have the least rearranged mt genomes among Acariformes mites
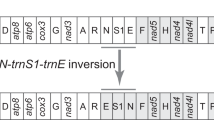
Like in other mites of the Acariformes, rearrangement of mt genes occurred in both of the eriophyoid mites. We calculated breakpoints with CREx as a measure of the extent of mt gene rearrangement from that of the hypothetical ancestor of arthropods36. Among the 12 different patterns of mt gene arrangement observed in the 27 species of Acariformes mites known, P. taishanensis has the least rearranged mt genome with 13 breakpoints and E. sabinae has the third least rearranged mt genome with 16 breakpoints (Table 2). In P. taishanensis, two rRNA genes (rrnS and rrnL) and seven tRNA genes (trnI, trnT, trnY, trnQ, trnV, trnW, trnM) changed their locations relative to their counterpart genes in the hypothetical ancestor of arthropods. In E. sabinae, two rRNA genes (rrnS and rrnL) and eight tRNA genes (trnK, trnI, trnT, trnY, trnQ, trnV, trnW, trnM) changed locations (Fig. 2). Four derived gene arrangements, trnS1-trnI-trnE, nad6-trnT-cob, trnY-trnQ-rrnS-trnV-rrnL and trnW-nad2-trnM-trnC, were found in both eriophyoid mites but not in any other mites; these novel gene arrangements were candidate synapomorphies (i.e. shared derived characters) for the eriophyoid mites.

Mitochondrial gene arrangements in the hypothetical ancestor of Acariformes, Phyllocoptes taishanensis and Epitrimerus sabinae.
Underlined genes are on the N-strand. Translocated or inverted genes are color-coded (blue: inversion and translocation; green: translocation; orange: inversion). rRNA genes are in grey; control regions are in black. Abbreviations of gene names are the same as in Fig. 1. Lines between genes indicate gene rearrangement. The positions of trnQ and trnY are unclear. Question marks after tRNA genes indicate uncertain locations.
Truncated tRNAs of the eriophyoid mites
Fifteen mt tRNA genes of P. taishanensis and 16 mt tRNA genes of E. sabinae were identified with tRNAscan-SE37 or ARWEN38 programs. The other seven tRNA genes of P. taishanensis (trnA, trnG, trnQ, trnR, trnS1, trnT, trnV) and the other six tRNA genes of E. sabinae (trnA, trnG, trnI, trnQ, trnR, trnS1) were found manually based on conserved nucleotides and the anticodon sequences. The putative mt tRNAs were highly truncated in both P. taishanensis (47 to 61 bp) and E. sabinae (47 to 67 bp) (Figs 3 and 4). Sixteen of the 22 tRNAs have atypical secondary structures, missing either D-arm or T-arm in both mites. The majority of tRNAs also have mismatches on T-arm, D-arm, acceptor arm or anticodon arm.

Inferred secondary structures of the 12 mt tRNAs of P. taishanensis (Pt) and E. sabinae (Es).
tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashes indicate Watson–Crick bonds; dots indicate bonds between U and G. Shared identical sequences between tRNA genes are circled in P. taishanensis.

Inferred secondary structures of the 10 mt tRNAs of P. taishanensis (Pt) and E. sabinae (Es).
tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashes indicate Watson–Crick bonds; dots indicate bonds between U and G. Shared identical sequences between tRNA genes are circled in P. taishanensis.
The phylogenetic position of Eriophyoidea
The super-family Eriophyoidea was traditionally in the order Trombidiformes. In our analysis, however, the two eriophyoid species were not grouped with spider mites (Tetranychidae), follicle mites (Demodicidae), chigger mites (Trombiculidae) and water mites (Unionicolidae), which were in the Trombidiformes. Rather, these two eriophyoid mites were grouped with Sarcoptiformes mites in ML and BI trees with strong support (Figures S1A-S1B, S1F, BSPs =100; Figure S2A, BPPs =100), or at the base in the Acariformes (Fig. 5, Figures S1C-S1E, Figures S2B-S2H). Excluding the two eriophyoid mites, the other Trombidiformes mites formed a monophyletic group with strong support (Table 3; Figures S1A-S1D, BSPs =100; Figures S2A, S2C-S2H, BPPs =100; Figure S2B, BPPs = 58). In addition, our phylogenetic analyses support the monophyly of Acariformes in all ML and BI trees (Table 3; Fig. 5, Figures S1A-S1F, BSPs >97; Figures S2A-S2I, BPPs =100). Monophyly of the other superorder, Parasitiformes, was recovered in ML and BI trees constructed with nucleotide sequences (Table 3; Figure S1A- S1D, S1F, BSPs >98; Figure S2A, S2C-S2I, BPPs =100), but was rejected in ML and BI trees constructed with amino acid sequences (Table 3, Figure S1E, Figure S2B). Also, three orders (Ixodida, Mesostigmata and Sarcoptiformes) were recovered as monophyletic groups in most of our analyses (Table 3, Fig. 5). Nine families (Acaridae, Argasidae, Demodicidae, Eriophyidae, Ixodidae, Phytoseiidae, Tetranychidae, Trombiculidae and Unionicolidae) were recovered as monophyletic groups in all of our analyses (Table 3, BSPs =100, BPPs =100). Intriguingly, monophyly of Acari was rejected as Pseudoscorpiones was grouped with Acariformes with strong support (Figure S1F, BSPs =98; Figure S2I, BPPs = 100). Our result thus conflicts with previous analyses based on rDNA in which Solifugae was grouped with Acariformes39,40,41, indicating the weaknesses of current hypotheses on the ordinal relationships in the Arachnida.

Maximum likelihood trees inferred with nucleotide sequences by 16 partitions (13 PCGs, 2 rRNA genes and concatenated tRNA genes).
Asterisks indicate the branches with >75% BSPs and>95% BPPs in the majority of the 13 topologies (Figures S1 and S2). Translocated genes are in green; inverted genes are in orange; inverted and translocated genes are in blue. rRNAs are in grey. Control regions are in black. Abbreviations of gene names are the same as in Fig. 1. Genes are transcribed from left to right except for those underlined, which are transcribed from right to left.
Discussion
The ancestral pattern of mt gene arrangement of Acariformes mites
Mitochondrial gene arrangement is relatively stable within major animal lineages8. This is, however, not the case for the Acari (ticks and mites). For the superorder Parasitiformes, the ancestral pattern of mt gene arrangement to arthropods was found in 11 species of Argasidae, one species of Allothyridae and six Ixodes species of Ixodidae (Fig. 5). Four Mesostigmata species and 16 Ixodidae species have rearrangement of mt genes (Fig. 5, Table 2). In the other superorder Acariformes, however, rearrangement of mt genes was found in all of the 27 species whose mt genomes have been sequenced. Even the species in the same genus differ from one other in mt gene arrangement, such as the two water mites in the genus Unionicola34,35 and three chigger mites in the genus Leptotrombidium11,42 (Fig. 5). The two eriophyoid mites investigated in the current study were from the same tribe (Phyllocoptini) of the family Eriophyidae but differ from one another in mt gene arrangement.
What was the ancestral pattern of mt gene arrangement for Acariformes mites ? Establishment of the ancestral pattern will lay the ground for understanding how mt genome organization evolved in Acariformes mites. Intriguingly, in comparison to other species of the Acariformes, the two eriophyoid mites have the least rearranged mt genomes and retained most of the ancestral pattern of mt gene arrangement of arthropods (Figs 2 and 5). The conserved mt gene-arrangement characters present in the two eriophyoid mites and other Acariformes mites allowed us to infer that the ancestral pattern of mt gene arrangement of the Acariformes retained likely the ancestral pattern of mt gene arrangement of arthropods with slight modifications (Fig. 2). The modifications include: 1) translocations of trnQ and trnY; these two tRNA genes are rearranged in all of the 27 species of Acariformes mites investigated to date and furthermore, their locations vary among different Acariformes lineages (Figs 2 and 5) inversion of rrnS-trnV-rrnL as a cluster, which is seen in the two eriophyoid mites and five Sarcoptiformes mites (Fig. 5); however, in seven of the 27 Acariformes species, rrnS and rrnL are translocated but not inverted, so there are alternative interpretations for the location and transcription orientation of rrnS-trnV-rrnL in the ancestor of Acariformes mites.
tRNA truncation in Acariformes mites
Truncated mt tRNAs have been found in several orders of the class Arachnida including Araneae25,26,27, Acariformes11, Pseudoscorpiones28, Scorpiones29,30 and Thelyphonida30. In the superorder Acariformes, truncated tRNAs are common and have been observed in all of the 27 species whose mt genomes have been sequenced. Sixteen of the 22 tRNAs in the two eriophyoid mites investigated in the current study lack D-arm or T-arm (Figs 3 and 4). Nineteen tRNAs lack D-arm or T-arm in spider mites (Tetranychidae); in some species, the tRNAs for phenylalanine and glutamine lack both D-arm and T-arm (Table S1)43,44. Fifteen tRNAs lost D-arm or T-arm in Demodicidae, including five tRNAs losing both D-arm and T-arm45. Twenty tRNAs in Pyroglyphidae46,47, 17 to 18 tRNAs in Trombiculidae11,34,42, 21 tRNAs in Acaridae and Psoroptidae48,49,50, 15 tRNAs in Unionicolidae34,35, lack D-arm or T-arm (Table S1).
Large-scale tRNA truncation was first observed and best studied in nematodes24. Twenty of the 22 tRNAs lack T-arm and the two tRNAs for serine lack D-arm in all nematodes except for several species in the class Enoplea. In Trichinella spiralis, eight tRNAs have the cloverleaf secondary structure with both D- and T-arms whereas several others lack both D- and T-arms51. tRNA truncation in the Acariformes mites has some similarity to that in nematodes but has their distinctive features. First, trnK has the typical cloverleaf structure in all Acariformes mites except for Steganacarus magnus9 (but K was not identified in this mite, see Table S1), whereas the other 21 tRNAs lack either D-arm or T-arm or both arms in one or more species of Acariformes mites. Three tRNAs, for cysteine, phenylalanine and histidine respectively, lack T-arm in all known Acariformes mites (Table S1), indicating that T-arm loss in these three tRNAs is likely ancestral to Acariformes mites. The other 18 tRNAs, however, vary in their secondary structures among the Acariformes mites: either having a cloverleaf structure, or lacking D-arm, T-arm or both arms (Table S1). The pattern of D-arm or T-arm loss appears to be consistent within a family but differs between families (Table S1). Thus, the loss of D-arm or T-arm in these tRNAs may have occurred multiple times independently in different lineages of the Acariformes mites.
Limited evidence from nematodes indicates that truncation does not seem to disable mt tRNAs from function. Okimoto et al. hybridized mt tRNA gene-specific probes to the RNAs of two nematodes, Caenorhabditis elegans and Ascaris suum and obtained evidence for the transcription of at least nine C. elegans and three A. suum mt tRNA genes52. Each tRNA transcript has the same size of its corresponding tRNA gene, to which three nucleotides (CCA) were added after transcription. Okimoto et al. concluded that the mt tRNAs without D-arm or T-arm were functional. Further, Suematsu et al. and Ohtsuki and Watanabe showed that the evolution of truncated mt tRNAs in nematodes was linked to EF-Tu protein: the paralogs of this protein acquired differential binding abilities to tRNAs with deleted domains53,54. Recently, Juhling et al. showed computational evidence that tRNAs of the nematodes of the class Enoplea that lack both D-arm and T-arm were functional51. The Acariformes mites provided another system for further experimental and computational investigation into the evolution and function of truncated mt tRNAs.
Should Eriophyoidea be placed in the order Trombidiformes?
The subclass Acari comprises two superorders, Acariformes and Parasitiformes55. The former includes two orders (Trombidiformes and Sarcoptiformes), while the later include four orders (Opilioacarida, Holothyrida, Ixodida, Mesostigmata)55. The monophyly of Acari, however, is not without controversy, when species of other major lineages of Arachnida were included into analyses28,40,41,56. Pseudoscorpiones was always grouped with Acariformes in our analyses, which is consistent with Ovchinnikov and Masta28. Several phylogenetic studies of Acariformes39,40,41 and Parasitiformes57 have been conducted previously using molecular data; however, eriophyoid mites were not included. The current taxonomic assignment of Eriophyoidea to the order Trombidiformes55 is controversial, due to the distinctive morphology of eriophyoid mites such as having only two pairs of legs, lack of ontogenetic diversity and absence of respirator system58. Indeed, André placed Eriophyoidea outside Trombidiformes59 Our analyses based on different partitions and inference methods showed consistently that Acariformes, Parasitiformes and several families (Acaridae, Argasidae, Demodicidae, Ixodidae, Phytoseiidae, Tetranychidae, Trombiculidae and Unionicolidae) were monophyletic, which was consistent with previous studies using mt genes9,18,27,39,44,45,46,50 or nuclear genes18,39,57. The monophyly of Trombidiformes, however, was rejected in our analyses (Figure S1, Figure S2). The monophyly of Trombidifromes was also rejected in a recent study by Gu et al. based on the mt genome sequences of 16 species of Acariformes mites due to spider mites (Tetranychidae) grouped with sarcoptiform mites50. In our analyses, when the two species of eriophyoid mites were excluded, the rest of the Trombidiformes species were always together in a monophyletic group. Our results thus raised the need for further phylogenetics studies on Acariformes mites with more taxa included especially from Tydeoidea and Eupodoidea, which were thought to be the sister groups of Eriophyoidea60. Further morphological studies are also necessary to elucidate the position of eriophyoid mites in Acariformes.
In conclusion, we sequenced the mt genomes of two species of eriophyoid mites, P. taishanensis and E. sabinae. These two mites have the least rearranged mt genomes seen in the Acariformes and have highly truncated mt tRNAs. Our comparison between the eriophyoid mites and other mites and ticks showed that the most recent common ancestor of Acariformes mites retained the ancestral pattern of mt gene arrangement of arthropods with slight modifications. The truncation of three tRNAs (for cysteine, phenylalanine and histidine, respectively) likely occurred once in the common ancestor of Acariformes mites. Truncation of other tRNAs, however, occurred multiple times independently in different lineages of Acariformes mites. Our phylogenetic analyses of mt genome sequences rejected the monophyly of the order Trombidiformes when eriophyoid mites were included. Further phylogenetics studies on Acariformes mites including more taxa from different lineages is needed to clarify the position of eriophyoid mites.
Methods
Collection of mites
E. sabinae and P. taishanensis were collected in May 2013 in Nanjing, China. E. sabinae was collected from Juniperus chinensis (Cupressaceae) (China savin), while P. taishanensis from Cedrus deodara (Pinaceae) (Deodar cedar). Mite samples were either used immediately for DNA extraction or were preserved in 100% ethanol at −20 °C prior to DNA extraction. Samples of each eriophyoid species were also mounted to slides as voucher, using modified Berlese medium61 for morphological check with Zeiss A2 (microphoto camera AxioCam MRc) microscope. All of the specimens and vouchers were deposited at the Arthropod Collection, Department of Entomology, Nanjing Agricultural University, China.
DNA extraction, mt genome amplification and sequencing
Genomic DNA was extracted from both individual and pooled specimens for each species, using a DNeasy Blood and Tissue Kit (QIAGEN), following the modified protocol62. For E. sabinae, a 658-bp fragment of cox1 and a 413-bp fragment of rrnL were initially amplified by PCR with the primer pairs LCO1490–HCO219863 and LRJ12287–LRN1339864 (see Additional file Table S2). PCR products were purified and sequenced directly using Sanger method at Majorbio (Shanghai, China). Specific primers for E. sabinae, ECOISR3 and E16SSR2, were designed from the sequences of the cox1 and rrnL fragments, respectively. PCR with these two primers produced a 1.7-kb amplicon, which was sequenced using Sanger method at Majorbio. Another pair of primers, PF2–PR2, were designed from the sequence of the 1.7-kb amplicon. An 11.2-kb amplicon was produced with PF2–PR2 primer pair and was sequenced with Illumina Hiseq 2000 platform at the Beijing Genome Institute, Hong Kong (BGI-HK).
For P. taishanensis, a 658-bp fragment of cox1 and a 407-bp fragment of rrnL were initially amplified by PCR with the primer pairs LCO1490-HCO219863 and LRJ12287–LRN1339864 (see Additional file Table S2). The PCR products were purified and sequenced directly using Sanger method at Majorbio. Two pairs of specific primers, TYCB3R2-TY16sR1 and RTYCB3F5-RTY16sF4, were designed from the sequences of the cox1 and rrnL fragments. The PCR with TYCB3R2-TY16sR1 produced a 1.7-kb amplicon, which was sequenced using Sanger method at Majorbio. The PCR with RTYCB3F5-RTY16sF4 produced an 11.2-kb amplicon, which was sequenced with Illumina Hiseq 2000 platform at the BGI-HK.
The initial PCRs contained 12.5 μL of PCR SuperMix (Transgen Biotech Co., Ltd., Beijing, China), 2 μl of template DNA and 1.25 μM of each primer, in a total volume of 25 μL. The PCR cycling conditions were: 3-min denaturation at 96 °C; 35 cycles of 10-sec denaturation at 95 °C, 30-sec annealing at 46 °C and 1.5-min extension at 72 °C; 5-min final extension at 72 °C; and then held at 4 °C. PCR products were checked on 1% agarose gel. PrimeSTAR GXL DNA polymerase (TAKARA) was used in the long PCRs with the cycling conditions: 98 °C for 10 sec, 68 °C for 2 to 10 min (depends on the length of regions between rrnL and cox1). The reaction mixture contained 0.5 μl GXL DNA Polymerase, 5 μl buffer, 2 μl dNTP mixture, 0.75 μl of each primer, 1 μl of template DNA and Milli-Q water added to total volume of 25 μl. Positive and negative controls were executed with each PCR. PCR products were checked on 1% agarose gel. PCR products were purified with QIAquick Spin PCR Purification Kit (QIAGEN).
Assembly of Illumina sequence-reads, gene identification and gene rearrangement analysis
Illumina sequence-reads obtained from the mt genome amplicons of P. taishanensis and E. sabinae were assembled into contigs with Geneious 6.1.6 (Biomatters Ltd.). The transfer RNA (tRNA) genes were identified using tRNAscan-SE37 and ARWEN38 or identified manually based on anticodons and secondary structures. tRNA genes of the two eriophyoid mites were verified by comparison of secondary structures and conserved nucleotide sequences with those of the Acari species reported in published literature. PCGs were identified by open reading frame search in Geneious and BLAST searches of GenBank65. The two rRNA genes, rrnL and rrnS, were also identified by BLAST searches of GenBank based on sequence similarity and conserved sequence motifs. The start and stop nucleotides of rrnL and rrnS cannot be determined exactly and were assumed to be immediately after their upstream genes and before their downstream genes. Breakpoints were calculated with CREx36 web server (http://pacosy.informatik.uni-leipzig.de/crex) as a measure of the extent of mt gene rearrangement; the two eriophyoid mites were compared with the hypothetical ancestor of arthropods (see Table 3 for details). The nucleotide sequences of mt genomes of P. taishanensis and E. sabinae have been deposited in GenBank under accession numbers KR604967 and KR604966.
Phylogenetic analyses
Sequences of the mt genomes of 64 Acari species, including 39 Parasitiformes mites10,11,12,13,14,15,16,17,18,19,20,21,22,66,67 and 25 Acariformes mites9,11,34,35,42,43,44,45,46,47,48,49,50,68 and two outgroup species were retrieved from GenBank (see Additional file Table S3). We used two horseshoe crabs, Limulus polyphemus69 and Carcinoscorpius rotundicauda, as outgroups. Horseshoe crabs have ancestral gene arrangement of arthropod and are in the same subphylum, Cheilcerata, as mites and ticks. To investigate the phylogenetic position of Acari in Arachnida, 18 more mt genomes were retrieved from GenBank, including two species of Amblypygi30, four species of Araneae26,30, two species of Opiliones30,70, three species of Scorpiones29,30, two species of Solifugae30,71, one species of Thelyphonida30, two species of Pseudoscorpiones28 and two species of Ricinulei71. Amino acid sequences of the PCGs were aligned individually using MAFFT v7.20572 web server (http://mafft.cbrc.jp/alignment/server/) with G-INS-i strategy for global homology and manually inspected before concatenation. Nucleotide sequences of PCGs were aligned using TranslatorX73 web server (http://translatorx.co.uk/) using MAFFT to compute the protein alignments. rRNA and tRNA genes were aligned individually using Muscle algorithm implemented in MEGA 6.0674; large gaps and ambiguous sites were deleted manually.
We analyzed the datasets of mt genome sequences as two types of matrix: amino acid sequences of PCGs and nucleotide sequences of all genes (13 PCGs, 2 rRNA genes and 22 tRNA genes). The datasets were partitioned by genes, by codon positions and optimal partitioning as determined by PartitionFinder (Table S4). Amino acid sequences were partitioned by 13 genes. Nucleotide sequences partitioned by genes resulted in 16 datasets: 13 PCGs, 2 rRNA genes and concatenated tRNA genes. Partitioning by codon positions resulted in six datasets: one for each base of codons, two for each rRNA gene and one for the combined tRNAs. Both types of partitioned nucleotide sequences were run twice independently, once with the third codon positions of PCGs included and once without the third codon positions. To avoid redundant sampling of each genus, which may potentially affect a complex lower-level phylogeny, two representative species from each genus (Argas, Ornithodoros, Amblyomma, Haemaphysalis, Ixodes, Rhipicephalus, Tetranychus, Leptotrombidium) were included in the analyses. The reduced datasets included 43 taxa and only Baysian inference was run based on the best models found by PartitionFinder. To check if RNA genes could affect the topology, we also ran Baysian inference with the nucleotide sequences of the 13 PCGs based on the best models found by PartitionFinder. The datasets with 18 additional Arachnid mt genomes were ran in ML and Baysian analyses with nucleotide sequences of the 13 PCGs by 13 partitions.
The best models of the datasets were predicted by jModelTest 2.1.175 and PartitionFinder v1.1.176, using the Bayesian Information Criterion (BIC). PartitionFinder was set using unlinked branch lengths, greedy search for nucleotide sequences and amino acid sequences. MtRev + I + G + F was chosen as the best amino acid substitution model for most PCGs, except for nad4, for which JTT + I + G + F was chosen as the best model. Nucleotide substitution model GTR + I + G was chosen as the best of 16 partitions. The ML analyses were performed using the GTRGAMMA model for nucleotide partitions and MtRev + I + G + F (JTT + I + G + F for nad4) for amino acid partitions in the program raxmlGUI1.377,78. For nodal support evaluation, a nonparametric bootstrap with 1,000 replicates was used. Mixed-model Bayesian analyses were performed with MrBayes 3.2.279 using separate data partitions. For BI, one cold chain and three heated chains were run with the combined dataset for 2 million generations. The average standard deviation of split frequencies fell down quickly. After 0.2 million generations, the average standard deviation number was below 0.01 in most of the BI trees. The convergence of the parameter estimates was performed with Tracer v1.6. The consensus three was edited with FigTree1.4.0.
Additional Information
How to cite this article: Xue, X.-F. et al. Mitochondrial genome evolution and tRNA truncation in Acariformes mites: new evidence from eriophyoid mites. Sci. Rep. 6, 18920; doi: 10.1038/srep18920 (2016).
References
Zhang, Z.-Q. in Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness (Magnolia Press, 2011).
Skoracka, A., Smith, L., Oldfield, G., Cristofaro, M. & Amrine, J. W. Host-plant specificity and specialization in eriophyoid mites and their importance for the use of eriophyoid mites as biocontrol agents of weeds. Exp. Appl. Acarol. 51, 93–113 (2010).
Duso, C., Castagnoli, M., Simoni, S. & Angeli, G. The impact of eriophyoids on crops: recent issues on Aculus schlechtendali, Calepitrimerus vitis and Aculops lycopersici. Exp. Appl. Acarol. 51, 151–168 (2010).
Castagnoli, M., Lewandowski, M., Labanowski, G. S., Simoni, S. & Soika, G. M. An insight into some relevant aspects concerning eriophyoid mites inhabiting forests, ornamental trees and shrubs. Exp. Appl. Acarol. 51, 169–189 (2010).
Miller, A. D. et al. Phylogenetic analyses reveal extensive cryptic speciation and host specialization in an economically important mite taxon. Mol. Phylogenet. Evol. 66, 928–940 (2013).
Navia, D. et al. Wheat curl mite, Aceria tosichella and transmitted viruses: an expanding pest complex affecting cereal crops. Exp. Appl. Acarol. 59, 95–143 (2013).
de Lillo, E. & Skoracka, A. What’s “cool” on eriophyoid mites ? Exp. Appl. Acarol. 51, 3–30 (2010).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Domes, K., Maraun, M., Scheu, S. & Cameron, S. L. The complete mitochondrial genome of the sexual oribatid mite Steganacarus magnus: genome rearrangements and loss of tRNAs. BMC Genomics 9, 532 (2008).
Jeyaprakash, A. & Hoy, M. A. The mitochondrial genome of the predatory mite Metaseiulus occidentalis (Arthropoda: Chelicerata: Acari: Phytoseiidae) is unexpectedly large and contains several novel features. Gene 391, 264–274 (2007).
Shao, R., Mitani, H., Barker, S. C., Takahashi, M. & Fukunaga, M. Novel mitochondrial gene content and gene arrangement indicate illegitimate inter-mtDNA recombination in the chigger mite, Leptotrombidium pallidum. J. Mol. Evol. 60, 764–773 (2005).
Burger, T. D., Shao, R., Labruna, M. B. & Barker, S. C. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick-borne Dis. 5, 195–207 (2014).
Mans, B. J., de Klerk, D., Pienaar, R., de Castro, M. H. & Latif, A. A. The mitochondrial genomes of Nuttalliella namaqua (Ixodoidea: Nuttalliellidae) and Argas africolumbae (Ixodoidae: Argasidae): estimation of divergence dates for the major tick lineages and reconstruction of ancestral blood-feeding characters. PLoS One 7, e49461 (2012).
Shao, R. et al. The mitochondrial genomes of soft ticks have an arrangement of genes that has remained unchanged for over 400 million years. Insect Mol. Biol. 13, 219–224 (2004).
Black, W. C. & Roehrdanz, R. L. Mitochondrial gene order is not conserved in arthropods: prostriate and metastriate tick mitochondrial genomes. Mol. Biol. Evol. 15, 1772–1785 (1998).
Shao, R., Barker, S. C., Mitani, H., Aoki, Y. & Fukunaga, M. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: a case study with Australasian Ixodes ticks. Mol. Biol. Evol. 22, 620–629 (2005).
Montagna, M. et al. Tick-box for 3’-end formation of mitochondrial transcripts in Ixodida, basal chelicerates and Drosophila. PLoS One 7, e47538 (2012).
Burger, T. D., Shao, R., Beati, L., Miller, H. & Barker, S. C. Phylogenetic analysis of ticks (Acari: Ixodida) using mitochondrial genomes and nuclear rRNA genes indicates that the genus Amblyomma is polyphyletic. Mol. Phylogenet. Evol. 64, 45–55 (2012).
Burger, T. D., Shao,. R. & Barker, S. C. Phylogenetic analysis of the mitochondrial genomes and nuclear rRNA genes of ticks reveals a deep phylogenetic structure within the genus Haemaphysalis and further elucidates the polyphyly of the genus Amblyomma with respect to Amblyomma sphenodonti and Amblyomma elaphense. Ticks Tick-borne Dis. 4, 265–274 (2013).
Navajas, M., Le Conte, Y., Solignac, M., Cros-Arteil, S. & Cornuet, J.-M. The complete sequence of the mitochondrial genome of the honeybee ectoparasite mite Varroa destructor (Acari: Mesostigmata). Mol. Biol. Evol. 19, 2313–2317 (2002).
Dermauw, W., Vanholme, B., Tirry, L. & Van Leeuwen, T. Mitochondrial genome analysis of the predatory mite Phytoseiulus persimilis and a revisit of the Metaseiulus occidentalis mitochondrial genome. Genome 53, 285–301 (2010).
Swafford, L. & Bond, J. E. The symbiotic mites of some Appalachian Xystodesmidae (Diplopoda: Polydesmida) and the complete mitochondrial genome sequence of the mite Stylochyrus rarior (Berlese) (Acari: Mesostigmata: Ologamasidae). Invertebr. Syst. 23, 445–451 (2010).
Wolstenholme, D. R. Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Wolstenholme, D. R., Macfarlane, J. L., Okimoto, R., Clary, D. O. & Wahleithner, J. A. Bizarre tRNAs inferred from DNA sequences of mitochondrial genomes of nematode worms. P. Natl. Acad. Sci. 84, 1324–1328 (1987).
Masta, S. E. & Boore, J. L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol. 21, 893–902 (2004).
Qiu, Y., Song, D., Zhou, K. & Sun, H. The mitochondrial sequences of Heptathela hangzhouensis and Ornithoctonus huwena reveal unique gene arrangements and atypical tRNAs. J. Mol. Evol. 60, 57–71 (2005).
Liu, M., Zhang, Z. & Peng, Z. The mitochondrial genome of the water spider Argyroneta aquatica (Araneae: Cybaeidae). Zool. Scr. 44, 179–190 (2014).
Ovchinnikov, S. & Masta, S. E. Pseudoscorpion mitochondria show rearranged genes and genome-wide reductions of RNA gene sizes and inferred structures, yet typical nucleotide composition bias. BMC Evol. Biol. 12, 31 (2012).
Dávila, S., Piñero, D., Bustos, P., Cevallos, M. A. & Dávila, G. The mitochondrial genome sequence of the scorpion Centruroides limpidus (Karsch 1879) (Chelicerata; Arachnida). Gene 360, 92–102 (2005).
Masta, S. E. & Boore, J. L. Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol. Biol. Evol. 25, 949–959 (2008).
Steinauer, M. L., Nickol, B. B., Broughton, R. & Ortí, G. First sequenced mitochondrial genome from the phylum Acanthocephala (Leptorhynchoides thecatus) and its phylogenetic position within Metazoa. J. Mol. Evol. 60, 706–715 (2005).
Beckenbach, A. T. & Joy, J. B. Evolution of the mitochondrial genomes of gall midges (Diptera: Cecidomyiidae): rearrangement and severe truncation of tRNA genes. Genome Biol. Evol. 1, 278–287 (2009).
Chen, W. J. et al. The mitochondrial genome of Sinentomon erythranum (Arthropoda: Hexapoda: Protura): an example of highly divergent evolution. BMC Evol. Biol. 11, 246 (2011).
Edwards, D. D., Jackson, L. E., Johnson, A. J. & Ernsting, B. R. Mitochondrial genome sequence of Unionicola parkeri (Acari: Trombidiformes: Unionicolidae): molecular synapomorphies between closely-related Unionicola gill mites. Exp. Appl. Acarol. 54, 105–117 (2011).
Ernsting, B. R., Edwards, D. D., Aldred, K. J., Fites, J. S. & Neff, C. R. Mitochondrial genome sequence of Unionicola foili (Acari: Unionicolidae): a unique gene order with implications for phylogenetic inference. Exp. Appl. Acarol. 49, 305–316 (2009).
Bernt, M. et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics 23, 2957–2958 (2007).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689 (2005).
Laslett, D. & Canback, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Dabert, M., Witalinski, W., Kazmierski, A., Olszanowski, Z. & Dabert, J. Molecular phylogeny of acariform mites (Acari, Arachnida): strong conflict between phylogenetic signal and long-branch attraction artifacts. Mol. Phylogenet. Evol. 56, 222–241 (2010).
Pepato, A. R., da Rocha, C. E. & Dunlop, J. A. Phylogenetic position of the acariform mites: sensitivity to homology assessment under total evidence. BMC Evol. Biol. 10, 235 (2010).
Pepato, A. R. & Klimov, P. B. Origin and higher-level diversification of acariform mites - evidence from nuclear ribosomal genes, extensive taxon sampling and secondary structure alignment. BMC Evol. Biol. 15, 178 (2015).
Shao, R., Barker, S. C., Mitani, H., Takahashi, M. & Fukunaga, M. Molecular mechanisms for the variation of mitochondrial gene content and gene arrangement among chigger mites of the genus Leptotrombidium (Acari: Acariformes). J. Mol. Evol. 63, 251–261 (2006).
Yuan, M.-L., Wei, D.-D., Wang, B.-J., Dou, W. & Wang, J.-J. The complete mitochondrial genome of the citrus red mite Panonychus citri (Acari: Tetranychidae): high genome rearrangement and extremely truncated tRNAs. BMC genomics 11, 597 (2010).
Chen, D. S. et al. The complete mitochondrial genomes of six species of tetranychus provide insights into the phylogeny and evolution of spider mites. PLoS One 9, e110625 (2014).
Palopoli, M. F., Minot, S., Pei, D., Satterly, A. & Endrizzi, J. Complete mitochondrial genomes of the human follicle mites Demodex brevis and D. folliculorum: novel gene arrangement, truncated tRNA genes and ancient divergence between species. BMC Genomics 15, 1124 (2014).
Dermauw, W., Van Leeuwen, T., Vanholme, B. & Tirry, L. The complete mitochondrial genome of the house dust mite Dermatophagoides pteronyssinus (Trouessart): a novel gene arrangement among arthropods. BMC Genomics 10, 107 (2009).
Klimov, P. B. & Oconnor, B. M. Improved tRNA prediction in the American house dust mite reveals widespread occurrence of extremely short minimal tRNAs in acariform mites. BMC Genomics 10, 598 (2009).
Sun, E. T., Li, C. P., Nie, L. W. & Jiang, Y. X. The complete mitochondrial genome of the brown leg mite, Aleuroglyphus ovatus (Acari: Sarcoptiformes): evaluation of largest non-coding region and unique tRNAs. Exp. Appl. Acarol. 64, 141–157 (2014).
Sun, E., Li, C., Li, S., Gu, S. & Nie, L. Complete mitochondrial genome of Caloglyphus berlesei (Acaridae: Astigmata): The first representative of the genus Caloglyphus. J. Stored Prod. Res. 59, 282–284 (2014).
Gu, X. B. et al. The complete mitochondrial genome of the scab mite Psoroptes cuniculi (Arthropoda: Arachnida) provides insights into Acari phylogeny. Parasite Vector 7, 340 (2014).
Jühling, F. et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 40, 2833–2845 (2012).
Okimoto, R. & Wolstenholme, D. A set of tRNAs that lack either the T psi C arm or the dihydrouridine arm: towards a minimal tRNA adaptor. The EMBO Journal 9, 3405 (1990).
Suematsu, T., Sato, A., Sakurai, M., Watanabe, K. & Ohtsuki, T. A unique tRNA recognition mechanism of Caenorhabditis elegans mitochondrial EF-Tu2. Nucleic Acids Res. 33, 4683–4691 (2005).
Ohtsuki, T. & Watanabe, Y.-I. T-armless tRNAs and elongated elongation factor Tu. IUBMB Life 59, 68–75 (2007).
Lindquist, E. E., Krantz, G. W. & Walter, D. E. in A manu of Acarology, third edition 8, 97–103 (Texas Tech University Press, 2009).
Dunlop, J. A. & Alberti, G. The affinities of mites and ticks: a review. J. Zoo. Syst. Evol. Res. 46, 1–18 (2007).
Klompen, H., Lekveishvili, M. & Black, W. C. Phylogeny of parasitiform mites (Acari) based on rRNA. Mol. Phylogenet. Evol. 43, 936–951 (2007).
Lindquist, E. E. & Amrine, Jr. J. W. in Eriophyoid Mites: Their Biology, Natural Enemies and Control 1, 33–87 (Elsevier, 1996).
Andre, M. Ordre des Acariens. Traité de zoologie 6, 794–892 (1949).
Lindquist, E. E. in Eriophyoid Mites: Their Biology, Natural Enemies and Control 1, 301–327 (Elsevier, 1996).
Amrine, Jr. J., W. & Manson, D. C. in Eriophyoid Mites: Their Biology, Natural Enemies and Control 1, 383–396 (Elsevier, 1996).
Dabert, J., Ehrnsberger, R. & Dabert, M. Glaucalges tytonis sp. n. (Analgoidea, Xolalgidae) from the barn owl Tyto alba (Strigiformes, Tytonidae): compiling morphology with DNA barcode data for taxon descriptions in mites (Acari). Zootaxa 1719, 41–52 (2008).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Simon, C. et al. Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Benson, D. A. et al. GenBank. Nucleic acids research 41, D36–D42 (2013).
Burger, T. D., Shao, R. & Barker, S. C. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol. Phylogenet. Evol. 76, 241–253 (2014).
Xu, Z. L. et al. Complete mitochondrial genome of Rhipicephalus simus. Mitochondr. DNA, early online : 1–2 (2014), doi: 10.3109/19401736.2014.919490.
Van Leeuwen, T. et al. Mitochondrial heteroplasmy and the evolution of insecticide resistance: non-Mendelian inheritance in action. P. Natl. Acad. Sci. USA. 105, 5980–5985 (2008).
Lavrov, D. V., Boore, J. L. & Brown, W. M. The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus. Mol. Biol .Evol. 17, 813–824 (2000).
Podsiadlowski, L. & Fahrei, K. The mitochondrial genome of Opilio parietinus (Arachnida: Opiliones). Mitochondr. DNA, 21, 149–150 (2010).
Fahrein, K., Talarico, G., Braband, A. & Podsiadlowski, L. The complete mitochondrial genome of Pseudocellus pearsei (Chelicerata: Ricinulei) and a comparison of mitochondrial gene rearrangements in Arachnida. BMC Genomics, 8, 386 (2007).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7–W13 (2010).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012).
Lanfear, R., Calcott, B., Ho, S. Y. & Guindon, S. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701 (2012).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Silvestro, D. & Michalak, I. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2011).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Acknowledgements
This research was funded by the National Natural Science Foundation of China (31172132), the Fundamental Research Funds for the Central Universities (KYZ201405), the State Scholarship Fund of China, the Australian Research Council (DP120100240 to RS) and Australia-China Science & Research Fund (ACSRF00980 to RS).
Author information
Authors and Affiliations
Contributions
X.-F.X., X.-Y.H. and R.S. designed the research. X.-F.X., J.-F.G. and Y.D. performed the research. X.-F.X. and R.S. analyzed the data. X.-F.X., X.-Y.H. and R.S. wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xue, XF., Guo, JF., Dong, Y. et al. Mitochondrial genome evolution and tRNA truncation in Acariformes mites: new evidence from eriophyoid mites. Sci Rep 6, 18920 (2016). https://doi.org/10.1038/srep18920
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18920
This article is cited by
-
The evolutionary characterization of Gamasida based on mitochondrial genes codon usage pattern
Parasitology Research (2024)
-
Symbiotic bacteria of the gall-inducing mite Fragariocoptes setiger (Eriophyoidea) and phylogenomic resolution of the eriophyoid position among Acari
Scientific Reports (2022)
-
Complete mitochondrial genomes of Thyreophagus entomophagus and Acarus siro (Sarcoptiformes: Astigmatina) provide insight into mitogenome features, evolution, and phylogeny among Acaroidea mites
Experimental and Applied Acarology (2022)
-
Mitochondrial analysis of oribatid mites provides insights into their atypical tRNA annotation, genome rearrangement and evolution
Parasites & Vectors (2021)
-
Genomic insights into mite phylogeny, fitness, development, and reproduction
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
