
Abstract
Monolayer Molybdenum Disulfide (MoS2) is a promising anode material for lithium ion batteries because of its high capacities. In this work, first principle calculations based on spin density functional theory were performed to investigate adsorption and diffusion of lithium on monolayer MoS2 with defects, such as single- and few-atom vacancies, antisite and grain boundary. The values of adsorption energies on the monolayer MoS2 with the defects were increased compared to those on the pristine MoS2. The presence of defects causes that the Li is strongly bound to the monolayer MoS2 with adsorption energies in the range between 2.81 and 3.80 eV. The donation of Li 2s electron to the defects causes an enhancement of adsorption of Li on the monolayer MoS2. At the same time, the presence of defects does not apparently affect the diffusion of Li and the energy barriers are in the range of 0.25–0.42 eV. The presence of the defects can enhance the energy storage capacity, suggesting that the monolayer MoS2 with defects is a suitable anode material for the Li-ion batteries.
Similar content being viewed by others

Introduction
With increasing demand for lithium ion batteries (LIBs) to be smaller and lighter, but still to have a higher energy density, extensive research is needed to find advanced electrode materials which can provide high specific capacity, long cyclic stability, high-rate capability and safety1,2. Higher capacities of 600 to 1000 mAh/g of mono-layer graphene and their composites3,4 compared to that of the bulk counterpart of graphite (372 mAh/g)5 inspired researchers to search for other monolayer anode materials for the LIBs, such as graphdiyne6, molybdenum disulfide (MoS2)7, boron carbon nitride nanosheets8 and monolayer V2O59, etc. MoS2 has a layered structure, in which the atoms are covalently bonded to form two-dimensional layers that are stacked together through weak van der Waals interactions10. The weak interactions between interlayers allow foreign ions or molecules to be introduced among the interlayers through intercalation without causing significant volume changes. Therefore, MoS2 could be developed as an intercalation host material to form a promising anode material for high energy density LIBs with a capacity of ~600 mAh/g11. Recently, nanostructured MoS2 has been attracted much attention for the anode of the LIBs and preliminary results showed it has a higher specific capacity than that of the bulk MoS212,13,14. The nanoflower MoS2 anode decorated with crumpled reduced graphene oxides exhibited a high specific capacity (1225 mAh/g) and an excellent cycling performance (680 mAh/g) after 250 cycles13. Ultrathin MoS2 nano-layers on N-doped carbon shells showed a high specific capacity of ~1000 mAh/g14. MoS2/graphene nanocomposite showed a high specific capacity of 1400 mAh/g in the first cycle and remained a specific capacity value of 1351 mAh/g after 200 cycles12.
Recently, two-dimensional (2D) MoS2 monolayer has been synthesized using different methods. It can be easily synthesized by a top-down methods by exfoliation from bulk materials, such as scotch tape based micromechanical exfoliation15,16, intercalation assisted exfoliation17,18,19 and liquid exfoliation20,21. The 2D MoS2 monolayer can also be synthesized using different bottom up approaches, such as transition metal sulfurization22,23, molybdenum oxide sulfurization24,25, physical vapor deposition26 and hydrothermal synthesis27,28. Defects are inevitably introduced during these fabrication processes and could significantly affect the physical, chemical and electrical properties of the two-dimensional MoS2 material. The presence of defects in the monolayer MoS2 has been proven experimentally and the types of defects were found to be dependent on the synthesized methods29,30,31,32. The dominant category of common defects could be changed from sulphur vacancy in the mechanical exfoliation and chemical vapor deposited samples to molybdenum antisite in the physical vapor deposited samples30,32. MoS2 monolayer with large areas could be synthesized using chemical vapor deposition (CVD)29. However, the obtained monolayer MoS2 is often polycrystalline in nature; thus dislocations and grain boundaries (GBs) normally appear in this monolayer23,31,33,34. The effects of intrinsic point defects (including various vacancies and antisite defects) and grain boundaries on the electronic and magnetic properties of the MoS2 monolayer have been investigated34,35,36.
The performance of the LIBs is significantly dependent on electrochemical properties of the cathode and anode materials37. The energy density is determined by the reversible capacity and operating voltage, which are generally determined by the electrode material chemistry, i.e., effective redox couples and maximum lithium concentration in active materials37. The rate capability and cycling performances are determined by the electronic and ion mobilities in the electrode materials. Apart from high energy density and fast ion mobility, electrode materials should be cheap and also have good thermal stability which is related to the safety of LIBs38. Theoretical studies revealed that the Li can be stably adsorbed onto the MoS2 monolayer with low diffusion barriers7,39. The intrinsic point defects (including various vacancies and antisite defects) and grain boundaries appear on the MoS2 monolayer, however, their effects on the Li adsorption remain unexplored. In this report, for the first time as far as we know, we perform density functional theory (DFT) calculations of Li adsorption and diffusion on pristine and MoS2 monolayers with various intrinsic point defects and grain boundaries. To be good anode materials for the LIBs, the diffusion barriers of the Li ion in the MoS2 should be small, which can realize a fast charging rate. At the same time, the MoS2 should have a large exothermic reaction energy with the lithium so that the anode materials have a large energy storage capacity.
Results
MoS2 monolayer has a sandwich-like structure with the Mo layer sandwiched between two layers of S. There are two polymorphs of MoS2, i.e., trigonal phase (1T) and hexagonal phase (2H). At room temperature, the MoS2 monolayer prefers to crystallize in a hexagonal phase19. The metal-stable 1T-MoS2 phase upon lithium and sodium intercalation has been observed by in-situ transmission electron microscopy (TEM) measurements40,41,42. The real mechanism of the 2H-1T phase transition is not well addressed. As discussed below, the defect formation energies in the metastable 1T-MoS2 have negative values, indicating that the 1T phase is not stable. Therefore, our investigations were focused mainly on the hexagonal phase structure. The calculated lattice parameters of monolayer 2H-MoS2 are: a = b = 3.22 Å. The Mo-S bond lengths have a constant of 2.448 Å, which is in a good agreement with experimental results43 and other simulation values44.
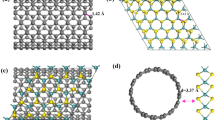
We considered the commonly observed point defects in the monolayer MoS230,32, including Mo (VMo) and S (VS) single vacancies, S2 double vacancies (VS2), a vacancy complex of Mo and nearby three sulfur (VMoS3), a vacancy complex of Mo and three nearby disulfur pairs (VMoS6) and antisite defects where a Mo atom substituting a S atom (MoS) or a S atom substituting a Mo atom (SMo). The optimized defect structures from the DFT calculations in the 2H-MoS2 and 1T-MoS2 are shown in Fig. 1(b,c), respectively. The defects of atomistic structures for all the point defects including VMo, VS, VS2, VMoS3, VMoS6 MoS and SMo in 2H-MoS2 show a 3-fold symmetry nature. Except for the point defects, an extended line defect GBs often appears in the samples prepared by a CVD method23,31,33,34. Based on direct mapping results of grain boundaries45,46 in the monolayer MoS2 using transmission electron microscope, the grain boundary is considered to be consisted of pentagon/heptagons (5–7) pairs with low energy configurations. The pentagon/heptagons (5–7) pairs are the basic components of GBs in the other layers materials, as evidenced by the TEM in graphene46,47. Therefore, we modeled the Li interaction with a grain boundary consisting of 5–7 polygons in the monolayers as shown in Fig. 1(a), with the GBs showing a mirror image symmetry. Each repeated grain boundary cell is composed of a Mo-rich structure with a homoelemental Mo-Mo bond and a reversed grain boundary with a sulfur-rich structure with two S-S bonds.

Atomistic configuration of MoS2 with defects.
Optimized atomistic configurations of (a) grain boundary and defects including VMo, VS, VS2, VMoS3, VMoS6, SMo in (b) 2H-MoS2 and (c) 1T-MoS2 monolayers.
Defects in MoS2 monolayer
The chemical potentials of Mo and S in equation (1) in a thermal equilibrium are defined by: (1) the values that prevent the formation of pure elements of Mo and S, in which the chemical potential cannot be higher than the energy of bulk materials of this species; (2) a thermal equilibrium with MoS2 chemical potential of Mo and S, which should satisfy the equation μMoS2 = μMo + 2μS. The defect formation energies as a function of S chemical potential, over a wide range of conditions from Mo-rich to S-rich for the 2H-MoS2, are shown in Fig. 2(a), where the 2H-MoS2 can remain stable with respect to the formation of bulk Mo (μS = −1.5 eV) or bulk alpha-S (μS = 0 eV). It can be seen that the VS has the lowest formation energy, which is consistent with the experimental observation that the VS is frequently observed30. VS2 is defined as a missing pair of S atoms aligned along the c-axis of the MoS2 lattice. The formation energy of the VS2 is about twice that of Vs, suggesting that the Vs does not tend to combine together. This can be supported by the fact that randomly distributed Vs was more frequently observed than the VS230. The formation energy of the VMo is higher than 4.59 eV even in the S-rich condition. Under the Mo-rich condition, VMoS3 has a lower defect formation energy than that of the VMo, which means that once the VMo is formed, the S atoms surrounding it become loose. The formation energy of large defects becomes higher as can be seen from Fig. 2(a) and the formation energy of VMoS6 is larger than that of VMoS3 for the whole chemical potentials. In the case of antisites, both the SMo and MoS have fairly low formation energy values for S-rich and Mo-rich conditions, respectively. The intrinsic defects can affect the electronic properties of the monolayer MoS2, thus much related work has been done to understand this effect30,32,35,44.

Defect formation energy.
Formation energies of different point defects as a function of sulfur chemical potential for (a) 2H-MoS2 in the range −1.5 eV < μS < 0 eV and (b) 1T-MoS2 in the range −0.95 eV < μS < 0 eV, where 2H-MoS2 can remain stable with respect to the formation of bulk Mo (μS = −1.5 eV) or bulk alpha-S (μS = 0 eV).
The defect formation energies as a function of S chemical potential over the range between Mo-rich and S-rich on 1T-MoS2 are shown in Fig. 2(b). All the defects have negative formation energies for the 1T-MoS2 in the whole S chemical potential range. The negative formation energy indicates that the defective 1T-MoS2 structures are more stable than the pristine ones. It is also reported that the critical value of lithium required for the stabilization of the 1T phase is estimated to x ≈ 0.4 in LixMoS248, now we will focus on how the defects affect the adsorption and diffusion the Li on the 2H-MoS2.
Adsorption of Li on 2H-MoS2 monolayer with point defects
There are two typical adsorption positions for a Li adatom on a pristine monolayer 2H-MoS2: a top site above a Mo atom (T site) and the hollow site in the hexagonal center (H site), as shown in Fig. 3. The calculated adsorption energies are −2.01 and −1.90 eV for the Li at T and H sites, respectively, which showed that the Li can stay in the T site more stable than that in the H site. This agrees well with the previously reported calculation results that the T site is the most stable site for Li adsorption39. The calculated adsorption energy for the Li adsorption on a pristine monolayer 2H-MoS2 at the T site is −2.01 eV with a Li-S bond length of 2.40 Å. All the possible positions for the Li adsorption around the point defects were calculated and the adsorption energies of the Li adsorbed onto different sites around the point defect are shown in Fig. 3, along with the lowest energy adsorption configurations. The absolute values of the adsorption energies of the Li on the defective 2H-MoS2 are increased compared with those on the pristine one. For example, the adsorption energy for the Li on the monolayer 2H-MoS2 with VMo is −3.54 eV, but this decreases to ~1.53 eV compared with the −2.01 eV for the Li adatoms on a pristine monolayer 2H-MoS2. The Li has favorable adsorption energy on the defective monolayer 2H-MoS2 than the pristine one. The strong binding energy leads to an enhanced capacity. In the presence of the VS and VS2, the most stable site for the Li adsorption is the bridge site between two Mo atoms on the S vacancy side. The Li adatom prefers to be adsorbed on the top site above the Mo vacancy when the VMo is presented in the monolayer 2H-MoS2, which is similar to the site of Li adsorbed on the pristine monolayer 2H-MoS2, but with a Li-S bond length of 2.32 Å. The Li adatom tends to be adsorbed on the bridge site between two Mo atoms as both the VMoS3 and VMoS6 are presented in the monolayer 2H-MoS2. The T site is still the most stable site for the Li adsorption as both the SMo and MoS are presented in the monolayer 2H-MoS2.

Adsorption energies and atomistic configurations.
The adsorption energies of Li adsorbed on different sites around the point defect and the most stable adsorption configurations.
The pristine monolayer 2H-MoS2 shows semiconductor properties. The valence band maximum (VBM) and conduction band minimum (CBM) are mainly composed of Mo 4d and S 3p states as seen from the projected density of states (PDOS) for both the Mo and S as shown in Fig. 4. As the Li atom is adopted on the pristine monolayer MoS2, the adsorption has no effect on the change of band gap and the near band edge band structure, except that the Fermi energy level moves upward into the conduction band. As the Li 2s state is ~3.7 eV, which means that Li is located above the CBM, the Li donates its electron to the CBM, thus pushing the Fermi level further upward. The Li adsorbed system shows an n-type doping state, which agrees well with the previous reports49. The electron transfer from the adatom to the substrate has been reported in other systems5,49,50,51. A charge transfer from K to the C layers is 0.57 electron for K adsorbed on graphene49. It was reported that a charge transfer of ~0.83 and 0.44 electron as Li atom adsorbed on the silicene51 and graphene50, respectively. The charge transfer will give rise to an n-type doping of the substrate49. Some peaks appear in the bandgap for the monolayer MoS2 with point defects, indicating that the point defects induce defect energy levels within the band gap. The Li 2s state locates at an energy higher than this defect levels and the Li donates the 2s electron to the point defects. Therefore, the Li ion has a positive charge and the defect has a negative charge and the adsorption was enhanced due to a strong coulomb interaction. The enhancement of Li adsorption in the defective graphene is also attributed to the same mechanism of coulomb interaction52.

Projected density of States.
Projected density of states of Li adsorbed on pristine monolayer MoS2 and the one with point defects.
Adsorption of Li on 2H-MoS2 monolayer with grain boundaries
The calculated formation energy of grain boundary is 0.20 eV/Å. The adsorption energies for a Li atom adsorbed at the different T and H sites of the monolayer 2H-MoS2 with a grain boundary are shown in Fig. 5. For most of the tested sites, the Li adatom prefers to occupy the T site rather than the H site in the monolayer MoS2 with the grain boundary. It can be seen that a Li adatom energetically prefers the adsorption sites near the grain boundary with a Mo-Mo bond, which indicates that the grain boundaries can significantly enhance the Li atom adsorption. For example, the Li adsorption energy at the T1 site is 0.33 eV, which is lower than that at the T12 site, indicating that Li is preferred to stay at the T1 site. The site projected density of states indicates that the localized states within the band gap come from the Mo and S atoms located at the grain boundaries. The adopted Li donates its electron to the localized states, thus increasing the adsorption between the Li ion and monolayer 2H-MoS2.

Li adsorption in MoS2 with GBs.
(a) Atomistic configuration of Li atom adsorbed at different T and H sites of the monolayer MoS2 with grain boundaries; (b) Dependence of the adsorption energy of Li adatom on different sites on monolayer MoS2 with grain boundaries; (c) Site projected density of states of monolayer MoS2 with grain boundaries.
Diffusion of Li on 2H-MoS2 monolayer with defects
As the charging rate of the LIBs relies on the Li ion mobility in the anode material, we studied the Li diffusion on the 2H-MoS2 monolayer with defects. As discussed above, VS has the lowest formation energy in all the S chemical potential over the range between Mo-rich and S-rich and SMo and MoS have fairly low formation energy values for S-rich and Mo-rich conditions, respectively. These defects have previously been confirmed from TEM observation30. We investigated the Li diffusion around the VS, SMo, MoS and GBs defects. The nudged elastic band method (NEB)53 is a good choice for calculating the energy barriers, yet it needs large computation efforts. We used a constrained method to calculate the diffusion barriers. The diffusion pathway of the Li atoms was determined by moving the Li atoms along different paths with a small constant distance; and the total energy was recorded at each position. The Li atom was constrained in the direction along the path, but it is free to move in the directions perpendicular to the path, which enable the Li atom to find its optimized position. As the stable adsorption site for the Li in pristine 2H-MoS2 monolayer is T site, the diffusion of Li occurs from one T site to a nearest T site by passing an H site, as shown in insert of Fig. 6(a). The calculated diffusion barrier for the Li in pristine 2H-MoS2 monolayer is 0.23 eV, which agrees well with NEB results of 0.2154 and 0.24 eV55. Clearly, the constrained method is reliable to be used to calculate the diffusion barrier of Li in 2H-MoS2 monolayer. Two diffusion paths were considered for the Li diffusion in 2H-MoS2 monolayer with VS defect and the energy curves as a function of relative diffusion coordinates are shown in Fig. 6(b,c). The VS defect does not affect the diffusion away from the defect; the energy barrier is 0.23 eV as Li diffuse from position 0 to 2. The energy barrier increases about 0.08 eV compared with the value in pristine MoS2 through path 1 in Fig. 6(b) and increases about 0.01 eV through path 2 in Fig. 6(c). The appearance of MoS defect does not significantly affect the diffusion behavior of Li in 2H-MoS2 monolayer, which can be concluded from Fig. 6(d). The SMo defect induces the increase of energy barriers up to about 0.09 eV. The energy barriers are kept at 0.23 eV as in the pristine 2H-MoS2 monolayer when the GBs appear. It can be seen from the energy curves in Fig. 6, the Li atom tends to diffuse to the defect position, therefore the diffusion barriers for backward diffusion of Li atoms were slightly increased. The maximum diffusion barriers are 0.37, 0.26, 0.42 and 0.30 eV for backward diffusion of Li atoms in 2H-MoS2 monolayer with VS, MoS, SMo and GBs defects, respectively. From these results we can conclude that the VS, MoS, SMo and GBs defects can enhance the adsorption of Li, but do not significantly affect the diffusion behavior of Li.

Diffusion of Li in MoS2 with defects.
Energy curves for Li ion diffusion on (a) pristine 2H-MoS2 monolayers and the one with (b)/(c)VS, (d) MoS, (e) SMo and (f) GBs defects.
Discussion
Defects will exist in real anode materials for LIBs, therefore, the effect of defects on the adsorption and diffusion Li in graphene52,56,57,58,59 and silicene60 has been investigated before. It was shown that the defects appear in graphene and silicene can enhance the adsorption of Li, thus can improve the Li storage capacities56. Together the results from this study, we can conclude that the presence of structural defects is beneficial for adsorption of Li atom in the two-dimensional materials. Li is bound to silicene with adsorption energies between 1.89 and 3.85 eV as the single vacancy, double vacancy and Stone-Thrower-Wales defects are presented60, which are higher than the values of 0.99–2.71 eV for the Li adopted on a defective grephene58. Our calculation shows that the Li can be strongly bounded to the defective 2H-MoS2 monolayer with adsorption energies in the range between 2.81 and 3.80 eV. The presence of the single-vacancy in graphene leads to a backward diffusion barrier of 0.56 eV52 and the presence of divacancy and Stone-Wales in the graphene leads to backward diffusion barriers of 0.37–0.54 eV58, which is higher than our calculated values of 0.26–0.42 eV for the Li in 2H-MoS2 monolayer with defects. The Li atom may be trapped by the defects in the graphene, thus cannot participate in the following electrochemical process. The presence of GBs in graphene leads to a decrease of about 0.92 eV in the adsorption energy of a Li adatom with diffusion barriers between 0.254 and 0.535 eV59. Our simulation results showed that the presence of GBs in the 2H-MoS2 monolayer leads a decrease of 1.2 eV in the adsorption energy of a Li adatom and keeps the low diffusion barrier of 0.23 eV as in the pristine 2H-MoS2 monolayer. As the adsorption energy is higher than that in the defective graphene and the diffusion barriers are lower than that in the graphene, MoS2 monolayer should be a better anode material for the LIBs compared to graphene.
Conclusion
The adsorption and diffusion of Li atom on the monolayer MoS2 with defects was studied using spin density functional theory. All the defects including single- and few-atom vacancies, antisite and grain boundary can enhance the adsorption of Li atom on the monolayer MoS2. The donation of Li 2s electron to the defects causes a strong coulomb interaction, thus enhances the adsorption. High adsorption energies and small diffusion barriers for the Li in the defective MoS2 suggested that a monolayer MoS2 with defects is a suitable anode material for the Li-ion batteries.
Simulations Details
All the calculations were performed using the spin density functional theory as implemented in the SIESTA code61. The electron exchange-correlation was processed using the generalized gradient approximation (GGA) with the parametrization scheme of Perdew-Burke-Ernzerhof (PBE)62 and the projector augmented wave (PAW) method62 was used to describe electron-ion interaction. Tests with a local density approximation (LDA) gave similar results for GGA in the calculations of the lattice parameters and band structures. Therefore, only the exchange-correlation potentials treated within GGA are applied for all the calculations. Electrons were described with norm-conserving Troullier-Martins pseudo-potentials63. The valence electron wave functions were expanded using double-ζ basis set plus polarization functions. The charge density was projected on a real space grid with a cutoff of 150 Ry to calculate the self-consistent Hamiltonian matrix elements.
A 5 × 5 hexagonal supercell of monolayer MoS2 was employed to model the point defects and the Li adsorption. A large spacing of 25 Å between the monolayers of MoS2 was used to prevent interlayer coupling. The Brillouin zone integration was modeled using a special k-point sampling of the Monkhorst-Pack scheme with a Г-centered grid. For the structural relaxation, a 3 × 3 × 1 k-grid was adopted for the calculations. All atomic positions and lattice constants were optimized using the conjugate gradient method until the maximum Hellmann-Feynman force acting on each atom was less than 0.02 eV/Å.
The defect formation energy Ef of defect α was calculated from the following expression64:

where E(α) is the total energy of the supercell containing a relaxed defect (vacancy, vacancy complex and antisite), E(pristine) is the total energy of the same supercell without defects, μi is the chemical potential of species i. ni is the number of exchanged particles between the supercell and the reservoirs in forming the defect cell. The formation energy of grain boundary was calculated using Ef = (Egrainbounary−Epristine)/LGB, where Egrainboundary and Epristine are the total energy values of a pristine MoS2 monolayer and the one with the grain boundary having the same number of MoS2 atoms pairs and LGB is the length of the grain boundary.
To analyze the stability of Li adsorbed on MoS2 with point defects, the adsorption energy of a Li adatom was calculated using

where  and
and  are the total energies of MoS2 with and without Li-adsorption, respectively.
are the total energies of MoS2 with and without Li-adsorption, respectively.  is the energy of an isolated lithium atom. According to the definition, a more negative binding energy indicates a more favorable exothermic reaction between the monolayer MoS2 and Li.
is the energy of an isolated lithium atom. According to the definition, a more negative binding energy indicates a more favorable exothermic reaction between the monolayer MoS2 and Li.
Additional Information
How to cite this article: Sun, X. et al. Defect-Mediated Lithium Adsorption and Diffusion on Monolayer Molybdenum Disulfide. Sci. Rep. 5, 18712; doi: 10.1038/srep18712 (2015).
References
Soloveichik, G. L. Battery technologies for large-scale stationary energy storage. Annu. Rev. Chem Biomol. 2, 503–527 (2011).
Liu, J. Addressing the grand challenges in energy storage. Adv. Funct. Mater. 23, 924–928 (2013).
Wang, G., Shen, X., Yao, J. & Park, J. Graphene nanosheets for enhanced lithium storage in lithium ion batteries. Carbon 47, 2049–2053 (2009).
Wang, D. et al. Ternary Self-Assembly of Ordered Metal Oxide-Graphene Nanocomposites for Electrochemical Energy Storage. ACS Nano 4, 1587–1595 (2010).
Dahn, J. R., Zheng, T., Liu, Y. H. & Xue, J. S. Mechanisms for Lithium Insertion in Carbonaceous Materials. Science 270, 590–593 (1995).
Sun, C. H. & Searles, D. J. Lithium Storage on Graphdiyne Predicted by DFT Calculations. J. Phys. Chem. C 116, 26222–26226 (2012).
Li, Y., Wu, D., Zhou, Z., Cabrera, C. R. & Chen, Z. Enhanced Li Adsorption and Diffusion on MoS2 Zigzag Nanoribbons by Edge Effects: A Computational Study. J. Phys. Chem. Lett. 3, 2221–2227 (2012).
Lei, W. W. et al. Large scale boron carbon nitride nanosheets with enhanced lithium storage capabilities. Chem. Commun. 49, 352–354 (2013).
Wang, Z., Su, Q. & Deng, H. Single-layered V2O5 a promising cathode material for rechargeable Li and Mg ion batteries: an ab initio study. Phys. Chem. Chem. Phys. 15, 8705–8709 (2013).
Silbernagel, B. G. Lithium intercalation complexes of layered transition metal dichalcogenides: An NMR survey of physical properties. Solid State Commun. 17, 361–365 (1975).
Feng, C. et al. Synthesis of molybdenum disulfide (MoS2) for lithium ion battery applications. Mater. Res. Bull. 44, 1811–1815 (2009).
Liu, Y. C., Zhao, Y. P., Jiao, L. F. & Chen, J. A graphene-like MoS2/graphene nanocomposite as a highperformance anode for lithium ion batteries. J. Mater. Chem. A 2, 13109–13115 (2014).
Xiong, F. Y. et al. Three-Dimensional Crumpled Reduced Graphene Oxide/MoS2 Nanoflowers: A Stable Anode for Lithium-Ion Batteries. Acs Appl. Mater. Inter. 7, 12625–12630 (2015).
Yu, X. Y., Hu, H., Wang, Y. W., Chen, H. Y. & Lou, X. W. Ultrathin MoS2 Nanosheets Supported on N-doped Carbon Nanoboxes with Enhanced Lithium Storage and Electrocatalytic Properties. Angew. Chem. Int. Edit. 54, 7395–7398 (2015).
Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. & Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 6, 147–150 (2011).
Lee, C. et al. Anomalous Lattice Vibrations of Single- and Few-Layer MoS2 . ACS Nano 4, 2695–2700 (2010).
Matte, H. et al. MoS2 and WS2 Analogues of Graphene. Angew. Chem. Int. Edit. 49, 4059–4062 (2010).
Zeng, Z. Y. et al. Single-Layer Semiconducting Nanosheets: High-Yield Preparation and Device Fabrication. Angew. Chem. Int. Edit. 50, 11093–11097 (2011).
Eda, G. et al. Photoluminescence from Chemically Exfoliated MoS2 . Nano Lett. 11, 5111–5116 (2011).
Coleman, J. N. et al. Two-Dimensional Nanosheets Produced by Liquid Exfoliation of Layered Materials. Science 331, 568–571 (2011).
Zhou, K. G., Mao, N. N., Wang, H. X., Peng, Y. & Zhang, H. L. A Mixed-Solvent Strategy for Efficient Exfoliation of Inorganic Graphene Analogues. Angew. Chem. Int. Edit. 50, 10839–10842 (2011).
Huang, X., Zeng, Z. Y., Fan, Z. X., Liu, J. Q. & Zhang, H. Graphene-Based Electrodes. Adv. Mater. 24, 5979–6004 (2012).
Lee, Y. H. et al. Synthesis of Large-Area MoS2 Atomic Layers with Chemical Vapor Deposition. Adv. Mater. 24, 2320–2325 (2012).
Seo, J. W. et al. Two-dimensional nanosheet crystals. Angew. Chem. Int. Edit. 46, 8828–8831 (2007).
Balendhran, S. et al. Atomically thin layers of MoS2 via a two step thermal evaporation-exfoliation method. Nanoscale 4, 461–466 (2012).
Lauritsen, J. V. et al. Size-dependent structure of MoS2 nanocrystals. Nat. Nanotechnol. 2, 53–58 (2007).
Peng, Y. Y. et al. Hydrothermal synthesis and characterization of single-molecular-layer MoS2 and MoSe2 . Chem. Lett. 772–773 (2001).
Peng, Y. Y. et al. Hydrothermal synthesis of MoS2 and its pressure-related crystallization. J. Solid State Chem. 159, 170–173 (2001).
Najmaei, S. et al. Vapour phase growth and grain boundary structure of molybdenum disulphide atomic layers. Nat. Mater. 12, 754–759 (2013).
Zhou, W. et al. Intrinsic Structural Defects in Monolayer Molybdenum Disulfide. Nano Lett. 13, 2615–2622 (2013).
Ly, T. H. et al. Observing Grain Boundaries in CVD-Grown Monolayer Transition Metal Dichalcogenides. ACS Nano 8, 11401–11408 (2014).
Hong, J. et al. Exploring atomic defects in molybdenum disulphide monolayers. Nat. Commun. 6, 6293 (2015).
Shi, Y. M. et al. van der Waals Epitaxy of MoS2 Layers Using Graphene As Growth Templates. Nano Lett. 12, 2784–2791 (2012).
Zhang, Z., Zou, X., Crespi, V. H. & Yakobson, B. I. Intrinsic Magnetism of Grain Boundaries in Two-Dimensional Metal Dichalcogenides. ACS Nano 7, 10475–10481 (2013).
Komsa, H.-P. & Krasheninnikov, A. V. Native defects in bulk and monolayer MoS2 from first principles. Phys. Rev. B 91, 125304 (2015).
Wang, Z. et al. Structure and electronic properties of transition metal dichalcogenide MX2 (M = Mo, W, Nb; X = S, Se) monolayers with grain boundaries. Mater. Chem. Phys. 147, 1068–1073 (2014).
Guo, Y. G., Hu, J. S. & Wan, L. J. Nanostructured materials for electrochemical energy conversion and storage devices. Adv. Mater. 20, 2878–2887 (2008).
Doi, T., Zhou, M. J., Zhao, L. W., Okada, S. & Yamaki, J. Influence of irreversible reactions at non-graphitizable carbon electrodes on their thermal stability in Li-ion batteries. Electrochem. Commun. 11, 1405–1408 (2009).
Chen, H. et al. Adsorption and Diffusion of Lithium on MoS2 Monolayer: The Role of Strain and Concentration. Int. J. Electro. Sci. 8, 2196–22003 (2013).
Wang, X., Shen, X., Wang, Z., Yu, R. & Chen, L. Atomic-Scale Clarification of Structural Transition of MoS2 upon Sodium Intercalation. ACS Nano 8, 11394–11400 (2014).
Wang, L., Xu, Z., Wang, W. & Bai, X. Atomic Mechanism of Dynamic Electrochemical Lithiation Processes of MoS2 Nanosheets. J. Am. Chem. Soc. 136, 6693–6697 (2014).
Cheng, Y. et al. Origin of the Phase Transition in Lithiated Molybdenum Disulfide. ACS Nano 8, 11447–11453 (2014).
Yang, D., Sandoval, S. J., Divigalpitiya, W. M. R., Irwin, J. C. & Frindt, R. F. Structure of single-molecular layer MoS2 . Phys. Rev. B 43, 12053–12056 (1991).
Ataca, C. & Ciraci, S. Functionalization of Single-Layer MoS2 Honeycomb Structures. J. Phys. Chem. C 115, 13303–13311 (2011).
Huang, P. Y. et al. Grains and grain boundaries in single-layer graphene atomic patchwork quilts. Nature 469, 389–395 (2011).
Kim, K. et al. Grain Boundary Mapping in Polycrystalline Graphene. ACS Nano 5, 2142–2146 (2011).
Huang, P. Y. et al. Grains and grain boundaries in single-layer graphene atomic patchwork quilts. Nature 469, 389–392 (2011).
Enyashin, A. N. & Seifert, G. Density-functional study of LixMoS2 intercalates (0 ≤ x ≤ 1). Comput. Theor. Chem. 999, 13–20 (2012).
Kaloni, T. P., Kahaly, M. U., Cheng, Y. C. & Schwingenschlogl, U. K-intercalated carbon systems: Effects of dimensionality and substrate. Europhys. Lett. 98, 67003 (2012).
Denis, P. A. Chemical Reactivity of Lithium Doped Monolayer and Bilayer Graphene. J. Phys. Chem. C 115, 13392–13398 (2011).
Kaloni, T. P., Schreckenbach, G. & Freund, M. S. Large Enhancement and Tunable Band Gap in Silicene by Small Organic Molecule Adsorption. J. Phys. Chem. C 118, 23361–23367 (2014).
Fan, X., Zheng, W. T. & Kuo, J.-L. Adsorption and Diffusion of Li on Pristine and Defective Graphene. ACS Appl. Mater. Interf. 4, 2432–2438 (2012).
Henkelman, G., Uberuaga, B. P. & Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Chen, H. J. et al. Adsorption and Diffusion of Lithium on MoS2 Monolayer: The Role of Strain and Concentration. Int. J. Electro. Sci. 8, 2196–2203 (2013).
Nasr Esfahani, D., Leenaerts, O., Sahin, H., Partoens, B. & Peeters, F. M. Structural Transitions in Monolayer MoS2 by Lithium Adsorption. J. Phys. Chem. C 119, 10602–10609 (2015).
Datta, D., Li, J., Koratkar, N. & Shenoy, V. B. Enhanced lithiation in defective graphene. Carbon 80, 305–310 (2014).
Yildirim, H., Kinaci, A., Zhao, Z.-J., Chan, M. K. Y. & Greeley, J. P. First-Principles Analysis of Defect-Mediated Li Adsorption on Graphene. ACS Appl. Mater. Interf. 6, 21141–21150 (2014).
Zhou, L.-J., Hou, Z. F. & Wu, L.-M. First-Principles Study of Lithium Adsorption and Diffusion on Graphene with Point Defects. J. Phys. Chem. C 116, 21780–21787 (2012).
Zhou, L.-J., Hou, Z. F., Wu, L.-M. & Zhang, Y.-F. First-Principles Studies of Lithium Adsorption and Diffusion on Graphene with Grain Boundaries. J. Phys. Chem. C 118, 28055–28062 (2014).
Setiadi, J., Arnold, M. D. & Ford, M. J. Li-Ion Adsorption and Diffusion on Two-Dimensional Silicon with Defects: A First Principles Study. ACS Appl. Mater. Interf. 5, 10690–10695 (2013).
Soler, J. M. et al. The SIESTA method for ab initio order-N materials simulation. J. Phys.-Conden. Matter. 14, 2745–2779 (2002).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Troullier, N. & Martins, J. L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 43, 1993–2006 (1991).
Van de Walle, C. G. & Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 95, 3851–3879 (2004).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (11474047). Funding support from Royal academy of Engineering UK-Research Exchange with China and India is acknowledged. This work was carried out at National Supercomputer Center in Tianjin and the calculations were performed on TianHe-1(A).
Author information
Authors and Affiliations
Contributions
The idea was conceived by Z.W. The simulation was performed by X.S. and Z.W. The data analyses were performed by X.S., Z.W. and Y.F. This manuscript was written by X.S., Z.W. and Y.F. All authors discussed the results and contributed to the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, X., Wang, Z. & Fu, Y. Defect-Mediated Lithium Adsorption and Diffusion on Monolayer Molybdenum Disulfide. Sci Rep 5, 18712 (2015). https://doi.org/10.1038/srep18712
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18712
This article is cited by
-
Phase evolution and structural modulation during in situ lithiation of MoS2, WS2 and graphite in TEM
Scientific Reports (2021)
-
A molecular dynamics study on the tribological behavior of molybdenum disulfide with grain boundary defects during scratching processes
Friction (2021)
-
Mechanical Properties of Two-Dimensional Materials (Graphene, Silicene and MoS2 Monolayer) Upon Lithiation
Journal of Electronic Materials (2020)
-
Two-dimensional black arsenic for Li-ion battery applications: a DFT study
Journal of Materials Science (2019)
-
Grain Boundaries Trigger Basal Plane Catalytic Activity for the Hydrogen Evolution Reaction in Monolayer MoS2
Electrocatalysis (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
