
Abstract
Herein, we report the use of a versatile photocleavable nitrobenzyl linker to micropattern a wide variety of bioactive molecules and photorelease them on demand. On one end, the linker has an NHS group that can be coupled with any amine, such as peptides, proteins or amine-linkers and on the other end an alkyne for convenient attachment to materials with an azide functional group. This linker was conjugated with NTA-amine or the cell adhesion peptide cRGD to enable straightforward patterning of His6-tagged proteins or cells, respectively, on PEGylated glass surfaces. This approach provides a practical way to control the presentation of a wide variety of bioactive molecules with high spatial and temporal resolution. The extent of photocleavage can also be controlled to tune the biomolecule density and degree of cell attachment to the surface.
Similar content being viewed by others
Introduction
The controlled presentation, patterning and release of bioactive molecules such as adhesion peptides and proteins on surfaces is important for the development of biomaterials that can guide cell behavior1. A key motivation to micropattern such bioactive molecules on surfaces and controllably detach them in vitro is to mimic the patterns and dynamics created during complex multicellular tissue formation in vivo. To turn on and off interactions dynamically, materials that react to stimuli, such as changes in temperature2, electric fields3,4,5, light6,7,8 or biochemical signals9,10,11, have been established1,12. Photoresponsive materials have been of particular interest as light provides external regulation with the highest accuracy in space and time, is non-invasive, can be used to pattern unconventional geometries (including both 2D and 3D), tune interactions and form gradients by only partial switching6,13,14.
The nitrobenzyl group, which undergoes selective bond cleavage upon irradiation with UV-light, has been a valuable building block to control material properties and the presentation of biomolecules6,13,14. Despite limitations of the nitrobenzyl group, which include slow reaction kinetics, low photochemical efficiency and irradiation with UV-light that can be damaging to cells, it has been used extensively to control cell material interactions6,13,14. To this end, the nitrobenzyl group has been integrated into linkers to attach protein-repellent PEG (poly ethylene glycol) coatings to different types of surfaces and to later locally release the coatings to obtain patches for cell adhesion15,16. The nitrophenethyl and nitrobenzyl photoremovable groups have also been used as a caging group on the amide back bone or the aspartic acid side chain of the cell adhesion peptide RGD (arginine glycine aspartic acid) to block integrin-mediated cell adhesion and to turn on cell adhesion upon illumination17,18,19. Conversely, the RGD motif has also been attached to PEG coatings through a nitrobenzyl linker to liberate the peptide upon illumination and render parts of the surface non-adhesive20. Further, nitrobenzyl groups have been introduced as crosslinkers into hydrogels to control their properties, degradation, the release of proteins from them and cell behaviour with light21,22,23. Such platforms have enabled the patterning of cells in confined and complex geometries and the study of the effect of geometric restrains on cell migration on glass substrates without introducing mechanical damage24,25. Additionally, the high temporal control provided by such light activated cell adhesion platforms has been exploited to study the role of cell adhesion in myogenic cell differentiation in culture26 and inflammation and vascularization in vivo27.
The patterning of proteins while preserving their activity is equally important for bioanalytical and biomedical applications. The binding of His6-tagged proteins to Ni2+-NTA (nitrilotriacetic acid) functionalized materials is a widely used approach for immobilization. Nitrobenzyl groups have been used to tune the interaction between His-tagged proteins and materials functionalized with Ni2+-NTA (nitrilotriacetic acid) and pattern His6-tagged proteins and protein complexes28,29,30,31. To achieve this the nitrobenzyl group has been incorporated into a photocleavable oligohistidine peptide backbone that can block the Ni2+-NTA complex on the surface and can be locally photocleaved for His6-tagged proteins to bind. While these examples provide an elegant ways to pattern His6-tagged proteins the synthesis of these peptides is challenging and it is not possible to release proteins from the surface. To overcome these limitations a complementary set-up where the Ni2+-NTA group can be cleaved from the surface selectively is desirable.
While all the above listed examples provide elegant platforms to micropattern adhesion peptides, cells and proteins, they are quite specific to a certain application. To facilitate the micropatterning of different bioactive molecules on materials and use them for cell studies, it is of interest to develop a generalized linker that can be easily conjugated to a wide variety of materials using click-chemistry and can be used to pattern a wide variety of bioactive molecules.
Results and Discussion
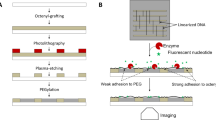
In this article, we present a versatile and convenient strategy to decorate materials with biologically active molecules through a nitrobenzyl linker that can be cleaved on demand. Considering the wide range of amine functional groups in bioactive molecules and the numerous materials with azide functional groups as points of attachment, we synthesized the nitrobenzyl linker 1 as reported in the literature16. As shown in Fig. 1a, the linker 1 can be photocleaved with UV light (365 nm), possesses an NHS-ester (N-Hydroxysuccinimide ester) functional group to react with an amine of choice and possesses an alkyne group for convenient attachment to any material with an azide functional group by using CuAAC (copper catalysed azide alkyne cycloaddition, also known as the “click reaction”). In previous reports linker 1 has been used to connect azide PEG molecules to amine-functionalized surfaces and release the PEG on demand to obtain the cell adhesive starting amine surface16. Here, however we envision to use linker 1 to attach a variety of bioactive molecules to passivating PEG coatings and liberate these to obtain inert surfaces. Most bioactive molecules and numerous linkers used for further functionalization methods possess amines, notably peptides, proteins and antibodies. To showcase the wide range of possible applications we coupled linker 1 to NTA-amine, a group that specifically interacts with His-tagged proteins or to the cell adhesion peptide cRGD (cyclic L-arginine glycine L-aspartic acid D-phenylalanine L-lysine), to yield NTA-NO2 and cRGD-NO2, respectively. Later, these molecules are immobilized on glass surfaces coated with a PEG-azide (a PEG3000 with an azide and a triethoxy-silane terminal group, Supplementary Fig. S1)32. The PEG coating is protein and cell repellent and the functionalization density with the conjugates of linker 1 can be tuned by adding non-reactive PEG2000 (a PEG2000 with a methoxy and a triethoxy-silane terminal group) during the coating reaction33. The patterning of the bioactive molecules is then realized by locally irradiating the surface in the geometry of interest and locally releasing the bioactive active molecule from the surface while the rest of the linker stays on the surface (Fig. 1b).

General functionalization strategy.
(a) Bioactive molecules with an amine such as cRGD and NTA-amine can be linked through the NHS moiety and the molecule can be attached to any material with an azide functional group through CuAAC. The nitrobenzene in the linker allows for the photochemical detachment. (b) Surfaces functionalized with bioactive molecules through linker 1 can be pattered by locally cleaving off the molecules with light.
As a first demonstration of our platform, we used the NTA-conjugate of linker 1, NTA-NO2, to pattern His6-tagged proteins (Fig. 2a). To characterize the photocleavage of the NTA-NO2 linker, we functionalized SiO2 QCM (quartz crystal microbalance) crystals with 10 or 100 mol% PEG-azide coatings, conjugated NTA-NO2 using the CuAAC to these and then loaded the NTA groups with NiCl2. Subsequently, the binding of His6-GFP to these surfaces and its release upon UV-irradiation was monitored in real time with QCM. It should be also noted that the UV-illumination causes an increase of a few Hz in the frequency, which is completely reversed when the illumination is stopped. In the case of crystals coated with 10 mol% PEG-azide, 75% of the previously bound His6-GFP is cleaved off after irradiation for 20 minutes and 60% of the His6-GFP is cleaved off from the surfaces with 100 mol% PEG-azide coating under the same conditions (Fig. 2b, Supplementary Fig. S2). It is possible to control the amount of protein release by controlling the exposure time to UV-light. From the QCM measurement it can be seen that about half of the bound His6-GFP is released form 10 mol% PEG-azide surfaces after 3 minutes of irradiation. The incomplete release of the protein could potentially be due to the low photochemical efficiency and slow kinetics observed for nitrobenzyl groups13,14. His6-GFP was also patterned on glass surfaces with the same set up. For this purpose a 100 mol% PEG-azide coated surface was modified as described above and illuminated under a microscope with a circular pattern (Fig. 2c). Later the surface was first incubated with NiCl2 and then His6-GFP. As can be seen from the intensity profile, His6-GFP does not bind to the illuminated areas but to the surrounding space (Fig. 2d). The only reported platform to photopattern His-tagged proteins is based on photocleavable oligohistidine peptides28,29,30,31. This approach selectively turns on protein binding to Ni2+-NTA sites upon illumination so that His-tagged proteins can be patterned in an oriented manner and allows for the patterning of multiple proteins, but the synthesis of such photocleavable peptides is rather complicated. Our approach is complementary to this existing platform, since we cleave the His-tag-binding Ni2+-NTA group from the material to achieve protein patterns. The linker can be photocleaved either before or after the binding of His6-tagged proteins to generate protein patterns. Therefore, this platform is not only useful to form protein micropatterns but also to photo-trigger protein release from these. This can potentially be used to study cell interactions with protein micropatterns and the uptake of immobilized proteins upon photo-cleavage.

Characterization of NTA-NO2.
(a) Working model of NTA-NO2. The His6-tagged POI (protein of interest) binds to the Ni2+-NTA-NO2 complex presented at the surface. Upon illumination the NTA group is cleaved from the surface and the protein is released. (b) His6-GFP binds on SiO2 QCM crystals coated with 10 mol% PEG-azide and modified with NTA-NO2. When the lamp is turned on the His6-GFP is liberated from the surface. (c) Fluorescence image of NTA-NO2 modified surface with circular micropatterns (160 μm) and (d) the line profile along the line. The surface was illuminated under an upright fluorescence microscope with an adjustable field aperture. The surface was incubated afterwards with Ni2+ and His6-GFP for visualization.
As a second case study, we conjugated linker 1 to the adhesion peptide cRGD to yield cRGD-NO2 (Fig. 3a). cRGD is recognized by the integrin αvβ3 on the cell surface and mediates cell adhesion. In QCM measurements integrin αvβ3 clearly binds to surfaces with 1 mol% PEG-azide coupled to cRGD-NO2 and the bound integrin dissociates completely from the surface upon irradiation with light (λ = 365 nm) for 20 minutes (Fig. 3b). In comparison, the binding of integrin αvβ3 is not reversed with irradiation when the peptide is linked through a non-photocleavable linker (Supplementary Fig. S3). This result excludes the possibility that the UV-light damages the coating and causes dissociation. In previous studies we showed that 1 mol% functionalization with cRGD is sufficient to sustain the adhesion and spreading of REF cells (rat embryonic fibroblast) on this type of PEG coatings32. Thus, we also used 1 mol% PEG-azide in all cell experiments to study cell adhesion behavior on cRGD-NO2 surfaces, which had been illuminated for various durations with a 365 nm light source (Fig. 3c). Before illumination as many REF cells adhere on the cRGD-NO2 surface as on the positive control surface with cRGD. Depending on the initial illumination time, the number of REF cells that can adhere to the cRGD-NO2 surface afterwards decreases gradually; if cells are seeded after 10 minutes of illumination only about half as many cells adhered to the surface as before illumination and after 30 minutes the number of cells that attached is comparable to the PEG-coating control. It also should be noted that the illumination of PEG-coated control surfaces did not impair the cell-repellent properties of the coating. Using cRGD-NO2 functionalized surfaces it is also possible to pattern adhesive and non-adhesive domains on the surface. Here, nonadhesive stripes are patterned under the microscope and MDCK (Madin-Darby canine kidney) epithelial cells are seeded on the surface (Fig. 3d). The MDCK cells only adhere on the stripes that were not illuminated and on the zones around the pattern but do not adhere on the illuminated regions.

Characterization of cRGD-NO2.
(a) Working model of cRGD-NO2. Cells can adhere to cRGD-NO2 modified PEG surfaces through integrin binding. Upon illumination the integrin ligand cRGD is cleaved off and cells can no longer adhere on the PEG coating. (b) QCM measurements showing integrin αvβ3 binding to cRGD-NO2 modified 1 mol% PEG-azide surfaces. The bound integrin αvβ3 is washed off as the surface is irradiated (λ = 365 nm). (c) REF cell adhesion on surfaces with 1% PEG-azide coating modified with cRGD-NO2. Surfaces are irradiated with light of λ = 365 nm for 0, 10 and 30 minutes to achieve various extends of cRGD cleavage. Unmodified surfaces with the same PEG coating are used as controls and the number of cells on the surface are given as the percentage of cells that adheres on a cRGD modified surface. The error bars are the standard deviation from three independent experiments. (d) Patterned MDCK cells on cRGD-NO2 modified surfaces. The surface was illuminated under an upright fluorescence microscope with an adjustable field aperture.
In summary, we demonstrated that linker 1 is a convenient tool to attach different bioactive molecules such as peptides and His6-tagged proteins to materials with azide functional groups as points of attachment and that these biomolecules can be cleaved on demand in a desired pattern non-invasively. While in this study we couple NTA-amine and cRGD to linker 1, this can be extended to many other biomolecules of interest that have amine functional groups such as antibodies, biotin-amine and proteins. The amount of bioactive molecules that are released from the surface can be tuned by controlling the illumination time (or light intensity) as is shown in QCM measurements and the cell adhesion experiments and this can potentially be used to form gradients of these biomolecules.
Methods
Synthesis of NTA-NO2
Linker 1 (1 eq., 19.6 mg, 50 μmol)16 dissolved in 850 μL DMF is added to Nα,Nα-bis(carboxymethyl)-L-lysine hydrate (1 eq., 13.2 mg, 50 μmol) dissolved in 850 μL 0.4 M of Na2CO3 pH 8.5 buffer. This mixture is kept at room temperature for 24 h and then lyophilized. The remaining solid is dissolved in water and extracted with ethyl acetate. The ethyl acetate phase is evaporated under vacuum and the remaining solid is purified by reverse phase HPLC (A: H2O, 0.1% TFA, B: acetonitrile, 0.1% TFA, gradient 20% to 80% B). HR-MS(ESI–): [M-2H+K]– observed: m/z = 576.11367, calc.: m/z = 576.12373. The concentration of the final product is determined to be 7 mM by UV-Vis (ε350 = 5000 M−1 cm−1)
Synthesis of cRGD-NO2
To a solution of linker 1 (2 eq., 2.6 mg, 6.6 μmol)16 in 120 μL DMF, a solution of cRGD (1 eq., 2.0 mg, 3.3 μmol) in 100 μL H2O and 60 μl of 1 M of Na2CO3 (pH 8.5) is added and the reaction mixture is kept at room temperature for 24 h. The reaction mixture is lyophilized and the remaining solid is purified by reverse phase HPLC (A: H2O, 0.1% TFA, B: Acetonitrile, 0.1% TFA, gradient 20% to 60% B). HR-MS(ESI+): [M+Na]+ observed: m/z = 903.36388, calc.: m/z = 903.36075. The concentration of the final product is determined to be 1.2 mM by UV-Vis (ε350 = 5000 M−1 cm−1)
Synthesis of PEG-azide
PEG-azide is synthesized following a similar protocol as described in previous publications for PEG200032,33. To a suspension of amino-terminated PEG-azide (azide-PEG3000-NH2, 1 eq., 500 mg, 1.67 mmol, Iris Biotech GmbH) in 1 ml DMF 3-(triethoxysilyl)-propylisocyanate (1.1 eq., 47.74 mg, 1.83 mmol) is added and the reaction is stirred under an argon atmosphere for 24 h at room temperature. The reaction solution is cooled to 0 °C, an excess of diethyl ether (10 ml) is added and the suspension is stirred for 1 h at 0 °C. The precipitate is filtered off, the product is washed with cold diethyl ether and the product is dried under vacuum. Yield: 540 mg, 1.66 mmol, ≈100% 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 3.89−3.28 (m, ≈350 H, OCH2 and NCH2), 3.17 (br s, 2 H, SiCH2CH2CH2), 1.68 (br s, 2 H, SiCH2CH2CH2), 1.25 (t, 9 H, CH3), 0.65 (br s, 2 H, SiCH2).
PEGylation of SiO2 surfaces
Glass slides (20 × 20 mm) are cleaned in freshly prepared Piranha solution (conc. 3:1 H2SO4:H2O2 (30%)) for 1 h, rinsed 3 times with Milli-Q water and dried in an N2 stream. The SiO2 coated QCM crystals are cleaned with a 2% SDS solution overnight, rinsed 3 times with Milli-Q water and dried in an N2 stream. The QCM crystals are then are treated with oxygen plasma (TePla 100-E, 0.4 mbar, 150 W, 10 min). For the PEGylation reaction, surfaces are immersed in a solution of appropriate PEG-azide and PEG2000 ratios (13.1 mg PEG2000 and 0.2 mg PEG-azide for 1% PEG-azide, 10.8 mg PEG2000 and 1.8 mg PEG-azide for 10% PEG-azide) and a drop of dry triethylamine in dry toluene (dried over molecular sieves (3 Å)) and kept at 80 °C overnight under a N2 atmosphere. The surfaces are first washed with ethyl acetate for 5 minutes by sonication, then with methanol for 5 minutes by sonication and dried in a N2 stream.
CuAAC Reaction on surfaces
The PEG-coated surfaces are incubated in contact with 50–100 μl of reaction solution containing 100 mM L-ascorbic acid, 100 mM Tris HCl (pH 8.5), 150 μM of the alkyne and 1 mM CuSO4 in a moisture chamber for 2 hours. Afterwards the surfaces are washed with water and dried under N2 stream. Alkynes used in this study include NTA-NO2 for QCM measurements and fluorescence measurements and cRGD-NO2 and cRGD alkyne for QCM measurements and cell adhesion studies. The surfaces functionalized with NTA-NO2 are washed with: 1) 50 mM EDTA (pH 7.4) for 5 min 2) Buffer A (50 mM Tris HCl (pH 7.4), 300 mM NaCl) for 5 min (2 times). 3) 0.1 M NiCl2 for 5 min. 4) Buffer A for 5 min. (2 times).
QCM (quartz-crystal microbalance) measurements
All QCM measurements are performed on a Q-Sense E4 system (Q-Sense) with SiO2 crystals (Q-sense) in a window module (Q-sense), which enabled in situ radiation with a 354 nm light source. All measurements are performed with a flow rate of 100 μl/min and at room temperature. QCM experiments performed with NTA-NO2 functionalized crystals consist of the following steps: 1) Buffer A as baseline (50 mM Tris HCl (pH 7.4), 300 mM NaCl), 2) 10 minutes 10 μM His6-GFP in Buffer A, 3) 10 minutes Buffer A, 4) 20 minutes UV-light with Buffer A, 5) 10 minutes Buffer A after UV exposure.
QCM experiments for investigating integrin αvβ3 binding to surfaces with cRGD-NO2 or cRGD and their photo-cleavage consist of the following steps: 1) Buffer B as baseline (50 mM Tris HCl (pH 7.4), 150 mM NaCl, 2 mM mgCl2, 1 mM MnCl2), 2) 30 minute incubation with 10 μg/ml integrin αvβ3 in Buffer B, 3) 10 minute Buffer B, 4) 20 minutes UV-light with Buffer B, 5) 10 minutes Buffer B after UV exposure.
Photopatterning
PEGylated surfaces clicked with NTA-NO2 (10:100 PEG-azide and PEG2000) or cRGD-NO2 (1:100 PEG-azide and PEG2000), are patterned using the Leica DM6000B microscope and a 20× air lens with the adjustable field aperture. Surfaces are exposed to light using the DAPI filter for 0.5–30 sec and then used for subsequent experiments. NTA-NO2 functionalized surfaces are: 1) Washed with buffer A, 2) incubated with 0.1 M NiCl2 for 5 minutes, 3) washed with buffer A for 5 minutes and 4) incubated with 10 μM His6-GFP for 20 minutes. Images are acquired with a Leica DM6000B under a 10× (HC PL APO 10×/0.40 Leica, Wetzlar, Germany) air lens. MDCK cells are seeded on cRGD-NO2 functionalized surfaces as described below.
Cell culture experiments
REF (rat embryonic fibroblasts) (kindly provided by B. Geiger, The Weizmann Institute of Science, Israel) and MDCK (Madin Darby Canine Kidney) epithelial cells are cultured in DMEM supplemented with 10% fetal bovine serum, 1% L-glutamine and 1% penicillin- streptomycin at 37 °C and 5% CO2. Surfaces used in cell experiments are washed with sterile PBS at room temperature and cells are cultured in serum-free medium during experiments on functionalized surfaces. Prior to seeding on the surfaces, cells are washed two times with 5 mL of sterile PBS, treated for 5 min with 2 mL of accutase at 37 °C and 5% CO2 and then resuspended in in serum-free medium. REF cells are seeded onto samples at a density of ca. 5000 cell/cm2 and incubated for 2 h at 37 °C and 5% CO2 before fixing the cells with 4% paraformaldehyde (PFA) in PBS (pH 7.4) for 20 min. Samples are washed twice with PBS and mounted in Mowiol containing DAPI (1 μg/mL). Fluorescence images are acquired on a Leica DM6000B microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Leica DFC 365 FX camera. A 10 × 10 tile scan is taken of each surface with a 10× (HC PL APO 10×/0.40 Leica, Wetzlar, Germany) air lens. The number of nuclei in the DAPI channel is analyzed with ImageJ 1.45s (http://imagej.nih.gov/ij) to calculate the number of cells per mm2 in comparison to a cRGD-coated surface. The error is given as the standard deviation of three independent experiments. MDCK cells are seeded at 3.5 × 105 cell/cm2 on to photopatterned surfaces and incubated for 2 hours at 37 °C and 5% CO2. The surfaces are washed twice with PBS to remove unattached cells and are then imaged.
Additional Information
How to cite this article: Wegner, S. V. et al. Photocleavable linker for the patterning of bioactive molecules. Sci. Rep. 5, 18309; doi: 10.1038/srep18309 (2015).
References
Kim, J. & Hayward, R. C. Mimicking dynamic in vivo environments with stimuli-responsive materials for cell culture. Trends Biotechnol. 30, 426–439 (2012).
Matsuda, N., Shimizu, T., Yamato, M. & Okano, T. Tissue engineering based on cell sheet technology. Adv. Mat. 19, 3089–3099 (2007).
Ng, C. C. A. et al. Using an electrical potential to reversibly switch surfaces between two states for dynamically controlling cell adhesion. Angew. Chem. Int. Ed. Engl. 51, 7706–7710 (2012).
Lamb, B. M. & Yousaf, M. N. Redox-switchable surface for controlling peptide structure. J. Am. Chem. Soc. 133, 8870–8873 (2011).
Yeo, W.-S., Yousaf, M. N. & Mrksich, M. Dynamic interfaces between cells and surfaces: electroactive substrates that sequentially release and attach cells. J. Am. Chem. Soc. 125, 14994–14995 (2003).
Cui, J., Miguel, V. S. & del Campo,A. Light-triggered multifunctionality at surfaces mediated by photolabile protecting groups. Macromol. Rapid Commun. 34, 310–329 (2013).
Auernheimer, J., Dahmen, C., Hersel, U., Bausch, A. & Kessler, H. Photoswitched cell adhesion on surfaces with RGD peptides. J. Am. Chem. Soc. 127, 16107–16110 (2005).
Liu, D., Xie, Y., Shao, H. & Jiang, X. Using azobenzene-embedded self-assembled monolayers to photochemically control cell adhesion reversibly. Angew. Chem. Int. Ed. Engl. 48, 4406–4408 (2009).
Liu, B., Liu, Y., Riesberg, J. J. & Shen, W. Dynamic presentation of immobilized ligands regulated through biomolecular recognition. J. Am. Chem. Soc. 132, 13630–13632 (2010).
Zhang, Z., Chen, N., Li, S., Battig, M. R. & Wang, Y. Programmable hydrogels for controlled cell catch and release using hybridized aptamers and complementary sequences. J. Am. Chem. Soc. 134, 15716–15719 (2012).
Boekhoven, J., Rubert Pérez, C. M., Sur, S., Worthy, A. & Stupp, S. I. Dynamic display of bioactivity through host-guest chemistry. Angew. Chem. Int. Ed. Engl. 52, 12077–12080 (2013).
Robertus, J., Browne, W. R. & Feringa, B. L. Dynamic control over cell adhesive properties using molecular-based surface engineering strategies. Chem. Soc. Rev. 39, 354–378 (2010).
Brieke, C., Rohrbach, F., Gottschalk, A., Mayer, G. & Heckel, A. Light-controlled tools. Angew. Chem. Int. Ed. Engl. 51, 8446–8476 (2012).
Klán, P. et al. Photoremovable protecting groups in chemistry and biology: reaction mechanisms and efficacy. Chem. Rev. 113, 119–191 (2013).
Nakanishi, J. et al. Photoactivation of a substrate for cell adhesion under standard fluorescence microscopes. J. Am. Chem. Soc. 126, 16314–16315 (2004).
Kaneko, S. et al. Photocontrol of cell adhesion on amino-bearing surfaces by reversible conjugation of poly(ethylene glycol) via a photocleavable linker. Phys. Chem. Chem. Phys. 13, 4051–4059 (2011).
Petersen, S. et al. Phototriggering of cell adhesion by caged cyclic RGD peptides. Angew. Chem. Int. Ed. Engl. 47, 3192–3195 (2008).
Wirkner, M. et al. Photoactivatable caged cyclic RGD peptide for triggering integrin binding and cell adhesion to surfaces. Chembiochem 12, 2623–2629 (2011).
Ohmuro-Matsuyama, Y. & Tatsu, Y. Photocontrolled cell adhesion on a surface functionalized with a caged arginine-glycine-aspartate peptide. Angew. Chem. Int. Ed. Engl. 47, 7527–7529 (2008).
Wirkner, M. et al. Triggered cell release from materials using bioadhesive photocleavable linkers. Adv. Mat. 23, 3907–3910 (2011).
Kloxin, A. M., Kasko, A. M., Salinas, C. N. & Anseth, K. S. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science 324, 59–63 (2009).
Kloxin, A. M., Tibbitt, M. W., Kasko, A. M., Fairbairn, J. A. & Anseth, K. S. Tunable hydrogels for external manipulation of cellular microenvironments through controlled photodegradation. Adv. Mater. 22, 61–66 (2010).
Azagarsamy, M. A. & Anseth, K. S. Wavelength-controlled photocleavage for the orthogonal and sequential release of multiple proteins. Angew. Chem. Int. Ed. Engl. 52, 13803–13807 (2013).
Rolli, C. G. et al. Switchable adhesive substrates: revealing geometry dependence in collective cell behavior. Biomaterials 33, 2409–2418 (2012).
Salierno, M. J., García, A. J. & del Campo, A. Photo‐activatable surfaces for cell migration assays. Advan. Funct. Mater. 23, 5974–5980 (2013).
Weis, S., Lee, T. T., del Campo, A. & García, A. J. Dynamic cell-adhesive microenvironments and their effect on myogenic differentiation. Acta Biomaterialia 9, 8059–8066 (2013).
Lee, T. T. et al. Light-triggered in vivo activation of adhesive peptides regulates cell adhesion, inflammation and vascularization of biomaterials. Nat. Mater. 14, 352–360 (2015).
Grunwald, C. et al. From the Cover: In situ assembly of macromolecular complexes triggered by light. Proc. Natl. Acad. Sci. USA 107, 6146–6151 (2010).
Gropeanu, M. et al. A versatile toolbox for multiplexed protein micropatterning by laser lithography. Small 9, 838–845 (2013).
Bhagawati, M., Lata, S., Tampé, R. & Piehler, J. Native laser lithography of His-tagged proteins by uncaging of multivalent chelators. J. Am. Chem. Soc. 132, 5932–5933 (2010).
Labòria, N., Wieneke, R. & Tampé, R. Control of nanomolar interaction and in situ assembly of proteins in four dimensions by light. Angew. Chem. Int. Ed. Engl. 52, 848–853 (2013).
Schenk, F. C., Boehm, H., Spatz, J. P. & Wegner, S. V. Dual-functionalized nanostructured biointerfaces by click chemistry. Langmuir 30, 6897–6905 (2014).
Bluemmel, J. et al. Protein repellent properties of covalently attached PEG coatings on nanostructured SiO(2)-based interfaces. Biomaterials 28, 4739–4747 (2007).
Acknowledgements
We thank the Max-Planck Society for financial support. J.P.S. is the Weston visiting professor at the Weizmann Institute for Science. The research group is part of the excellence cluster CellNetworks at the University of Heidelberg. S.V.W. thanks the Baden-Württemberg Stiftung for a postdoctoral fellowship with the Eliteprogramm. O.I.S. thanks the Erasmus Program for a fellowship.
Author information
Authors and Affiliations
Contributions
S.V.W. and J.P.S conceived the project and designed the experiments. S.V.W. and O.I.S. performed the experiments. S.V.W wrote the manuscript and prepared the figures.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wegner, S., Sentürk, O. & Spatz, J. Photocleavable linker for the patterning of bioactive molecules. Sci Rep 5, 18309 (2016). https://doi.org/10.1038/srep18309
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18309
This article is cited by
-
Current hydrogel advances in physicochemical and biological response-driven biomedical application diversity
Signal Transduction and Targeted Therapy (2021)
-
The increasing dynamic, functional complexity of bio-interface materials
Nature Reviews Chemistry (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
