
Abstract
Febrile seizures (FS) are the most common seizure syndrome and are potentially a prelude to more severe epilepsy. Although zinc (Zn2+) metabolism has previously been implicated in FS, whether or not variation in proteins essential for Zn2+ homeostasis contributes to susceptibility is unknown. Synaptic Zn2+ is co-released with glutamate and modulates neuronal excitability. SLC30A3 encodes the zinc transporter 3 (ZNT3), which is primarily responsible for moving Zn2+ into synaptic vesicles. Here we sequenced SLC30A3 and discovered a rare variant (c.892C > T; p.R298C) enriched in FS populations but absent in population-matched controls. Functional analysis revealed a significant loss-of-function of the mutated protein resulting from a trafficking deficit. Furthermore, mice null for ZnT3 were more sensitive than wild-type to hyperthermia-induced seizures that model FS. Together our data suggest that reduced synaptic Zn2+ increases the risk of FS and more broadly support the idea that impaired synaptic Zn2+ homeostasis can contribute to neuronal hyperexcitability.
Similar content being viewed by others


Introduction
Febrile seizures (FS) are the most common seizure syndrome, affecting 2–3% of children in the pre-school years1. FS account for over 1 in 200 paediatric emergency department (ED) admissions manifesting in physical, psychological and behavioural issues2. They may also be associated with more severe forms of epilepsy in later life with long-term studies indicating that 7% of children with FS subsequently develop epilepsy3. Despite the clinical burden little progress in understanding the causes of FS has been made over the last decade, making this area a key research priority for the epilepsy field4.
Several studies support the idea that low zinc (Zn2+) levels increase seizure susceptibility. For example, altering dietary Zn2+ intake can alter seizure susceptibility in a genetic mouse model of epilepsy, with low Zn2+ increasing sensitivity and high Zn2+ being protective5. Furthermore, rats administered intraperitoneal injections of the Zn2+ chelator sodium diethyldithiocarbamate develop seizures6. Importantly, Zn2+ levels are significantly lower in blood and/or cerebrospinal fluid of children that suffer FS; both when compared to healthy controls and when compared to children either presenting with fever alone or seizures not associated with fever7,8,9,10. These studies highlight dysfunction of Zn2+ homeostasis as a potential mechanism of enhanced FS susceptibility.
Genetic factors play an important role in determining FS susceptibility11,12. However, whether or not genetic variation in proteins essential for Zn2+ homeostasis contributes to FS susceptibility is not known. Zn2+ transporter 3 (ZNT3), encoded by SLC30A3, is well placed to modulate neuronal excitability. ZNT3 is primarily responsible for the transport of Zn2+ into synaptic vesicles where it is co-localised with glutamate and released in an activity-dependent manner13,14. High Zn2+ concentrations can occur in the extracellular space potentially regulating pre- and post-synaptic membrane excitability by modulating a variety of ion channels, receptors and transporters15. Synaptic Zn2+ released during short trains of activity inhibits NMDA receptors and hence acts as an important inhibitor of hippocampal neuronal circuit excitability14. Consistent with this, ZnT3 knock-out mice display increased susceptibility to pharmacological pro-convulsants16. Thus reduction in synaptic Zn2+ may increase neuronal excitability and consequently seizure susceptibility. Based on the central role of synaptic Zn2+ in modulating hippocampal excitability and clinical evidence implicating low cerebrospinal fluid and blood levels in FS we hypothesised that variation in ZNT3 would contribute to FS susceptibility. To address this we took a candidate gene approach, screened SLC30A3, and functionally validated a variant enriched in FS patients.
Results
ZNT3 sequencing reveals a R298C variant enriched in FS patients
Our screen of FS probands for variants in the coding and splice site regions of SLC30A3, encoding human ZNT3, revealed a variant (c.892C > T; p.R298C) in the cytoplasmic domain (Fig. 1a,c). This variant is enriched in FS probands (n = 3/286; 1%) and absent from population-matched controls (n = 0/643, P < 0.05, Fisher test). Two probands were from Australia (Probands 1 and 2) and one from Belgian (Proband 3). The variant is reported at over 10-fold lower frequency amongst the 6,491 exomes on the Exome Variant Server (0.09%; EVS; http://evs.gs.washington.edu/EVS/) and the 60,386 exomes on the Exome Aggregation Database (~0.08%; ExAC; http://exac.broadinstitute.org/).

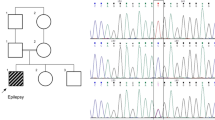
Enrichment of SLC30A3 variant in FS.
(a) Sequence chromatogram showing the c.892C > T SLC30A3 variant enriched in FS patients. (b) Multiple species alignment of ZNT3 protein sequence showing the R298 amino acid is highly conserved (arrow). Rhesus = Rhesus monkey; Prairie = Prairie vole. (c) Schematic showing domain structure of the ZNT3 protein. Light-blue shading indicates domains involved in Zn2+ binding. The R298C variant is located in the cytoplasmic domain near the C-terminus. Adapted from40.
The case-control analysis of this variant in FS probands against the EVS gives an odds ratio of 11 (CI 2–37). Given a lifetime prevalence of 2–3%, the 10 fold increase in risk implied by this odds ratio would lead to an absolute risk of over 1 in 5 of developing FS. High Grantham (180) and PolyPhen-2 (0.995/1) scores also suggest that the variant is probably damaging to the ZNT3 protein. Furthermore, the amino acid change occurs in a highly conserved region of the cytoplasmic domain (Fig. 1b,c) and SLC30A3 has low tolerance to variation (intolerance score = −0.8; 12th percentile)17. The variant substitutes a positively charged, polar arginine residue at position 298 with a cysteine residue possessing a thiol side chain that is susceptible to oxidation and may form disulphide bonds with other cysteine residues.
Inheritance of the R298C variant
All three probands had simple FS. A twin cohort made up a significant component of the sample (37/259) and probands 1 and 2 both have a monozygotic twin who has the R298C allele. The twin of proband 1 is unaffected while the twin of proband 2 had afebrile focal dyscognitive seizures in childhood (Fig. 2). Both inherited the variant from an unaffected parent. Proband 3 inherited the variant (Fig. 2) from her father, who was affected with febrile seizures intermixed with afebrile seizures until the age of 14 (febrile seizures plus, FS+). A brother was also affected with FS while there are another 4 unaffected carriers of the allele. Overall, the phenotypes in the relatives strengthen the argument that R298C is a risk allele rather than a Mendelian dominant allele and suggest an association with epilepsies wider than simply FS. Marker analysis was performed on the three probands and available relatives using microsatellites at the SLC30A3 locus (Supplementary Table 1). This revealed a small region of between 1.84 Kb and 1.50 Mb shared by all three probands (1, 2 and 3) and a larger region of between 1.03 Mb and 1.76 Mb shared by only the two Australian probands (1 and 2).

Pedigrees of probands carrying ZNT3 variant.
All three probands carry the c.892C > T (p.R298C) variant in SLC30A3. The segregation of the variants in their respective pedigrees is shown: *heterozygous c.892C > T (p.R298C) genotype (C/T); wt, homozygous wild-type genotype (C/C).
ZNT3 (R298C) causes a trafficking deficit resulting in severe loss-of-function
We developed a functional assay based on an established method in which ZNT3 is transfected into rat pheochromocytoma (PC12) cells18. Endogenous ZNT3 is present at only low level in PC12 cells and transfected ZNT3 protein traffics to synaptic-like microvesicle (SLMV) membranes providing a robust system to assay transporter function. To validate the SLMV enrichment method untransfected PC12 cells were fractionated by differential centrifugation to separate nuclear, endosomal/mitochondrial and SLMV fractions19. The three fractions were lysed and equal concentrations of protein from each fraction used, separated by SDS PAGE and Western blotted. Ponceau staining was used to verify the equivalence of protein concentration and transfer before probing with anti-synaptophysin1 and anti-v-glut1 antibodies. Signal was strongest in the SLMV fraction consistent with micro-vesicle enrichment (Fig. 3ai).

Functional analysis reveals a loss-of-function due to the ZNT3 (R298C) variant. ai.
Western blot analysis of the three fractions isolated by differential centrifugation including: cell/nuclear (Nucl.), endosomal/mitochondrial (Endo) and synaptic –like microvesicle (SLMV) fractions. Cross-reactivity with synaptophysin (SYP) and vGlut1 antibodies is greatest in the P3 (200,000 g) fraction confirming SLMV enrichment. aii Confocal images of representative PC12 cells transfected with eGFP, ZNT3 (WT)-eGFP and ZNT3 (R298C)-eGFP. Scale bar = 15 μm (b) Median eGFP fluorescence in PC12 cells transfected with ZNT3 (WT) and ZNT3 (R298C) confirming similar ZnT3 translation from both constructs (p = 0.74, n = 4, Student’s paired t-test).(c) Normalized Zn2+ concentration in the SLMV fraction of ZNT3 (WT) and ZNT3 (R298C) transfected PC12 cells from four independent experiments. Zn2+ was significantly reduced in ZNT3 (R298C) SLMVs (*p = 0.0013, n = 4, Student’s ratio paired t-test) (d) Mean fluorimetry measurements of eGFP in the SLMV fraction of ZNT3 (WT) and ZNT3 (R298C) transfected and in non-transfected PC12 cells. Fluorescence was significantly reduced in ZNT3 (R298C) relative to ZNT3 (WT) and was indistinguishable from auto-fluorescence in non-tranfected cells (*p = 0.0001, n = 3 for all groups, One way ANOVA).
In four independent experiments, duplicate flasks of PC12 cells were transiently transfected with either wild-type ZNT3 (WT) or mutant ZNT3 (R298C) both tagged with eGFP (Fig. 3aii). To overcome low transfection efficiency, whole cells expressing eGFP were sorted using FACS. Mean eGFP fluorescence per cell was equal for ZNT3 (WT) and ZNT3 (R298C) suggesting similar translation of both proteins (Fig. 3b). The isolated whole PC12 cells containing eGFP were mechanically lysed and the cell contents fractionated by differential centrifugation to obtain the SLMV enriched fraction. Zn2+ concentration was directly measured in the SLMV using mass spectrometry and showed a significantly lower Zn2+ concentration in cells transfected with ZNT3 (R298C) when compared to ZNT3 (WT) transfected cells (Fig. 3c). These results suggest a loss of transporter function.
eGFP-tagging provides an opportunity to measure the amount of ZNT3 protein present in the SLMV fraction using fluorimetry. In the SLMV fraction a significantly lower fluorescence signal was evident for ZNT3 (R298C) compared with ZNT3 (WT) (Fig. 3d). ZNT3 (R298C) fluorescence in the SLMV fraction was equivalent to a similar fraction isolated from untransfected cells suggesting an almost complete loss of trafficking of the mutant protein to the vesicle membrane (Fig. 3d).
ZnT3 null mice have heightened susceptibility to heat-mediated seizures
Genetic mouse models of epilepsy based on human FS mutations are more sensitive to heat-induced seizures validating the environmental heating assay20,21,22,23. Wild-type and ZnT3 knock-out mice do not differ in gross weight ruling out this confound as a potential basis of differential seizure susceptibility (6.5 ± 0.2 g WT, n = 8 vs. 6.5 ± 0.6 g, KO, n = 11, p > 0.95). Kaplan-Meier survival curve analysis shows that ZnT3 null mice develop heat-mediated clonic-tonic seizure in a significantly shorter time than wild-type mice (Fig. 4). Similarly, average time to clonic-tonic seizure was significantly faster for ZnT3 null mice (694 ± 73 s WT, n = 8 vs. 510 ± 37 s, KO, n = 11, p = 0.03). This is consistent with low synaptic Zn2+ levels increasing susceptibility to FS.

ZnT3 null mice have heightened sensitivity to heat-induced clonic-tonic seizures.
Kaplan-Meier curves showing time to first tonic-clonic seizure during exposure to heat for wild-type and ZnT3 knock-out mice homozygous are significantly different (p = 0.01, wild-type, n = 8; ZnT3, n = 11; Mantel-Cox test).
Discussion
We identify the ZNT3 (R298C) variant and show it to be enriched in human FS populations. Although the association does not reach genome wide significance, functional analysis in PC12 cells revealed the variant causes loss-of-function through a trafficking deficit. This loss-of-function is highly likely to predispose to FS as mice null for ZnT3 protein have a heightened sensitivity to thermogenic seizures. Collectively this data strongly implicate ZNT3 as a susceptibility gene in FS and more generally Zn2+ homeostasis as a critical modulator of neuronal excitability.
Genetic factors play an important role in FS susceptibility with about 25% of FS patients having a family history12. Furthermore, monozygotic twins have a higher FS concordance than dizygotic twins, strongly implicating a genetic component11. The familial epilepsy syndrome most closely associated with FS is genetic epilepsy with febrile seizures plus (GEFS+). FS+ are distinct from FS because they occur outside the usual 6 months to 6 years age range and are often intermixed with afebrile seizures24. In families with GEFS + mutations have been identified in sodium channel subunit genes, most commonly SCN1A but also SCN9A and SCN1B, as well as the GABA receptor subunit GABRG2 and the presynaptic protein syntaxin 1B (STX1B)25,26,27,28,29.
For the more common simple FS, much less of the genetic contribution is known. A genome-wide association study showed a number of common variant associations with FS, all with odds ratios under 230. Two SNPs were replicated in the study, one in the known epilepsy gene SCN1A and another in a transmembrane protein of as yet unclear function, ANO3. Two further SNPs were associated overall but not replicated, one in SCN2A and the other in a non-coding region implicated as a quantitative trait loci for serum Mg2+ levels. Furthermore, a quantitative trait locus-based study of sensitivity to environmental-heat induced seizures in mice found an association between seizure susceptibility and Srp9, which encodes a cytoplasmic ribonucleoprotein complex31. Subsequent analysis in humans showed an association of a SRP9 promoter region SNP with both FS and temporal lobe epilepsy31. Variation in Fgf13, HCN2, and KCC2 have also been implicated in FS32,33,34,35. Our findings add to these reports indicating that a variant in ZNT3 causing loss-of-function acts as a risk allele of moderate effect rather than a SNP of small effect or a dominant Mendelian allele.
The cellular assays confirm that the R298C variant leads to a marked loss of ZNT3 function. Given the equivalent translation of both ZNT3 (WT) and ZNT3 (R298C) proteins in PC12 cells, the lack of expression of ZNT3 (R298C) in the SLMV fraction points to a specific trafficking deficit. ZNT3 forms homodimers which are critical for the correct subcellular localization18. Interestingly, Salazar and colleagues showed that reducing dimerization of ZNT3 reduced trafficking to the SLMV fraction. Site-directed mutagenesis isolated the carboxy-terminus as a critical regulator of dimerization18. The ZNT3 (R298C) variant falls within the carboxy-terminus and is therefore well placed to disrupt dimerization, although future studies are required to determine the molecular basis of the trafficking deficit.
Multiple potential mechanisms may underlie increased excitability as extracellular Zn2+ interacts with a range of ion channels, receptors and transporters15,36,37,38. NMDA receptors, however, stand out as a target for Zn2+ because of their high sensitivity with levels as low as 10 nM producing significant inhibition under specific conditions39. Indeed, activity-dependent release of Zn2+ at hippocampal synapses modulates NMDA-mediated excitability14. This impact on excitability was limited to higher stimulation frequencies, a situation likely to occur during neuronal hyperexcitability induced by fever40. Here we show that ZnT3 knock-out mice are more sensitive to heat-induced seizures, a phenomenon we have previously shown to model a human FS phenotype. Importantly, Vergnano and colleagues demonstrated that trains of evoked NMDA receptor-mediated synaptic events are significantly larger in the ZnT3 knock-out mouse14. We propose a model of pathogenesis in which heat-mediated increases in hippocampal neuronal excitability are not constrained in patients harbouring ZNT3 (R298C) due to reduced inhibition of NMDA receptors by Zn2+.
Environmental Zn2+ deficiency also appears to predispose to FS. Several clinical studies report low blood and CSF Zn2+ levels in children who have been diagnosed with FS7,8,9,10. Our results provide the first genetic evidence that disruption in Zn2+ homeostasis can lead to an increased FS susceptibility. Interestingly, da Rocha and colleagues reported that serum Zn2+ levels appear to be influenced by a common SLC30A3 variant, although the mechanistic basis of this is unclear41. This cements the idea that low Zn2+ levels confer FS susceptibility through both environmental and genetic means.
Materials and Methods
Clinical Evaluation and Patient Sample
All experimental protocols were approved by The Human Research Ethics Committee of Austin Health, Melbourne, Australia and the Commission for Medical Ethics of the University of Antwerp, Antwerp, Belgium. All methods were carried out in accordance with the approved guidelines of The Human Research Ethics Committee of Austin Health, Melbourne, Australia and the Commission for Medical Ethics of the University of Antwerp, Antwerp, Belgium. Informed consent was obtained from all human subjects. A comprehensive assessment of each patient was obtained using a validated seizure questionnaire, clinical evaluation by an experienced epileptologist and review of relevant medical records and clinical investigations. Where possible patients also underwent neurological and general medical examination. Previous clinical records and reports of relevant investigations were obtained if available. We studied 286 unrelated patients of European ancestry recruited from Australia or Belgium diagnosed with FS and 643 control individuals without FS from the same populations. Whole blood was obtained and genomic DNA extracted using a Qiagen QIAamp DNA Maxi Kit (Valencia, CA).
PCR, Sanger Sequencing and Marker Analysis
The SLC30A3 gene was amplified using gene-specific primers (oligonucleotides available on request) designed to the full-length reference human gene transcript (NM_003459; NCBI Gene http://www.ncbi.nlm.nih.gov/). Amplification reactions were cycled using a standard protocol on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA) at 60 °C annealing temperature for 1 minute. Bidirectional sequencing of all exons and flanking regions was completed with a BigDyeTM v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer’s instructions. Sequencing products and microsatellite markers were resolved using a 3730xl DNA Analyzer (Applied Biosystems). All sequencing chromatograms were compared to published cDNA sequence; nucleotide changes were detected using Codon Code Aligner (Codon Code Corporation, Dedham, MA).
DNA constructs and mutagenesis
A clone of human SLC30A3 (ZNT3) CDS (NCB1 XM_006712100) in pUC 57 was obtained from GenScript (Piscataway, NJ, USA). PCR was used to introduce the variant into this construct and the construct was tagged at the carboxy-terminal with eGFP. Tagged constructs were inserted into a pCDNA 3.1 vector. DNA for transfection was assessed for integrity and quantitated by both Nano-drop 2000 (Thermo Scientific) and gel electrophoresis. OD260/280 ratios were 1.85–1.86. For transfection DNA was diluted to 1 ug/ul in sterile water.
Cell Culture
Pheochromocytoma (PC12) cells were grown and maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal bovine serum (FBS), 5% horse serum and 1% Penicillin-Streptomycin and held at 37 oC in 5% CO2. Cells were transfected by electroporation (AMAXA Cell Line Nucleofector kit V, Lonza, Basel, Switzerland). After 72 hours they were exposed to 25 μM ZnSO4 (in DMEM) and incubated for 1 hour followed by 3 washes in phosphate-buffered saline (PBS). Cells were detached enzymatically (Accutase, Sigma Aldrich, Australia), pelleted and resuspended in cell-sorter medium (PBS supplemented with 2% FBS and 1.5 mM EDTA). Non-transfected cells were used as a control. A 3 × 106 cells/ml−1 suspension was prepared for flow cytometry. Transfected cells were then separated by Fluorescence Activated Cell Sorting (FACS) using a BD Aria 111 cell sorter (BD Biosciences, San Jose, CA, USA).
Cell fractionation
FACS sorted cells were pelleted and resuspended in intracellular buffer (150 mM NaCl, 10 mM HEPES pH 7.4, 1 mM MgCl2 and Complete protease inhibitors (Roche, Basel, Switzerland)) and lysed by sonication for 1 second. Cell lysis was confirmed by microscopy. The lysates were fractionated by differential centrifugation as described previously19. Briefly, the lysed cells were centrifuged at 1000 g for 15 minutes to generate a nuclear pellet and the supernatant subsequently centrifuged at 27,000 g for 45 minutes, generating an endosomal/mitochondrial pellet with the final supernatant centrifuged at 200,000 g for 90 minutes to obtain a synaptic–like microvesicle (SLMV) enriched fraction. The SLMV enriched fractions were stored at −80 °C.
Imaging
Transfected PC12 cells were fixed in 2% paraformaldehyde for 20 minutes and mounted for imaging analysis. Confocal images were obtained using a Zeiss LMS 800 (Zeiss, Jena, Germany) microscope using excitation and emission wavelengths optimised for eGFP.
Zn2+ Analysis
Detergent lysed SLMV’s, from equal numbers of fractionated cells, were digested in concentrated high purity nitric acid (Aristar; BDH, London, UK) overnight at room temperature and then at 90 °C for 20 minutes. Samples were diluted with 1% nitric acid and measurements made using a Varian UltraMass inductively coupled plasma mass spectroscopy (ICPMS) instrument (PaloAlto, CA ,USA) under operating conditions suitable for routine multi-element analysis. The instrument was calibrated using blank, 10, 50 and 100 ppb of a certified multi-element ICPMS standard solution (ICP-MS-CAl2-1; AccuStandard) for Mn2+, Fe2+, Cu2+ and Zn2+ in 1% nitric acid. Results were obtained from four independent experiments. Mn2+ and Fe2+ were below levels of detection and Cu2+ values were inconsistent across the experimental groups. Zn2+ concentrations were untransfected < transfected mutant < transfected wild-type in all experiments. Zn2+ in un-transfected cells was deducted from the values in transfected cells prior to analysis. Comparisons between wild-type and mutant were made within experiments, with the wild-type value set as 1.
Fluorimetry of SLMV fraction
ZNT3 levels in the samples were assayed in duplicate samples by eGFP fluorimetry in a black-walled 384 well plate in the Polarstar Omega Plate Reader (BMG Labtech, Offenburg, Germany). Protocol parameters were: excitation 485–12, emission 520 and gain set at 3077. eGFP protein in lysis buffer was used to generate a standard curve to ensure readings fell in an assayable range.
Western blotting
Western blotting was carried out according to a previously published method42. Protein concentration in samples was assayed by the Bradford method (BioRad, Hercules, CA, USA) and equal protein concentrations from each fraction (80 μg) were separated by electrophoresis on a 12% SDS poly-acrylamide gel, transferred to nitrocellulose by semi-dry Western blotting and the filter stained with Ponceau S (Sigma, St Louis MO, USA) to confirm successful, even and equivalent protein transfer. Anti-synaptophysin1, 1:3000 (Synaptic Systems, Goettingen, Germany) and anti-vglut1, 1:500 (ab 104899, Abcam, Cambridge UK) primary antibodies and anti-rabbit HRP conjugated 1:3000 (BioRad, Hercules, CA, USA) and anti-sheep/goat HRP conjugated 1:1000 (Silenus, Amrad, Melbourne, Australia) secondary antibodies were used to confirm SLMV enrichment.
Thermogenic Seizure Testing
All animal experimentation was performed in accordance with ethics approval obtained from the Florey Institute of Neuroscience and Mental Health Animal Ethics Committee. Post-natal 16–17 mice were placed in a container heated to constant 41–42 °C and the time to first tonic-clonic seizure recorded20. Mice were sacrificed immediately after the first observed seizure to comply with our animal ethics approval. Comparisons were made between ZNT3 knock-out and their wild-type littermates43.
Statistical analysis
Statistical analysis for association was Fisher’s Exact Test calculated in R (version 2.10, http://R-project.org). Survival curves for each group were analysed using the Mantel-Cox method (GraphPad, CA, USA). Analysis for average time to seizure was completed using an unpaired t-test (GraphPad). Statistical analysis for the functional study used paired t-tests and one way ANOVA as appropriate, see Figure legends (GraphPad). P < 0.05 was set as significant. Graphs are mean ± standard error of the mean.
Additional Information
How to cite this article: Hildebrand, M. S. et al. Loss of synaptic Zn2+ transporter function increases risk of febrile seizures. Sci. Rep. 5, 17816; doi: 10.1038/srep17816 (2015).
References
Hauser, W. A., Annegers, J. F. & Rocca, W. A. Descriptive epidemiology of epilepsy: contributions of population-based studies from Rochester, Minnesota. Mayo Clin Proc 71, 576–586 (1996).
Martindale, J. L., Goldstein, J. N. & Pallin, D. J. Emergency department seizure epidemiology. Emerg Med Clin North Am 29, 15–27 (2011).
Annegers, J. F., Hauser, W. A., Shirts, S. B. & Kurland, L. T. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 316, 493–498 (1987).
Helbig, I. & Lowenstein, D. H. Genetics of the epilepsies: where are we and where are we going? Curr Opin Neurol 26, 179–185 (2013).
Fukahori, M. & Itoh, M. Effects of dietary zinc status on seizure susceptibility and hippocampal zinc content in the El (epilepsy) mouse. Brain Res 529, 16–22 (1990).
Blasco-Ibanez, J. M. et al. Chelation of synaptic zinc induces overexcitation in the hilar mossy cells of the rat hippocampus. Neurosci Lett 355, 101–104 (2004).
Burhanoglu, M., Tutuncuoglu, S., Coker, C., Tekgul, H. & Ozgur, T. Hypozincaemia in febrile convulsion. Eur J Pediatr 155, 498–501 (1996).
Ganesh, R., Janakiraman, L. & Meenakshi, B. Serum zinc levels are low in children with simple febrile seizures compared with those in children with epileptic seizures and controls. Ann Trop Paediatr 31, 345–349 (2011).
Salehiomran, M. R. & Mahzari, M. Zinc status in febrile seizure: a case-control study. Iran J Child Neurol 7, 20–23 (2013).
Waqar Rabbani, M., Ali, I., Zahid Latif, H., Basit, A. & Rabbani, M. A. Serum Zinc Level in Children Presenting with Febrile Seizures. Pak J Med Sci 29, 1008–1011 (2013).
Eckhaus, J. et al. Genetics of febrile seizure subtypes and syndromes: a twin study. Epilepsy Res 105, 103–109 (2013).
Kjeldsen, M. J., Kyvik, K. O., Friis, M. L. & Christensen, K. Genetic and environmental factors in febrile seizures: a Danish population-based twin study. Epilepsy Res 51, 167–177 (2002).
Qian, J. & Noebels, J. L. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol 566, 747–758 (2005).
Vergnano, A. M. et al. Zinc dynamics and action at excitatory synapses. Neuron 82, 1101–1114 (2014).
Marger, L., Schubert, C. R. & Bertrand, D. Zinc: an underappreciated modulatory factor of brain function. Biochem Pharmacol 91, 426–435 (2014).
Cole, T. B., Robbins, C. A., Wenzel, H. J., Schwartzkroin, P. A. & Palmiter, R. D. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res 39, 153–169 (2000).
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S. & Goldstein, D. B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9, e1003709 (2013).
Salazar, G., Falcon-Perez, J. M., Harrison, R. & Faundez, V. SLC30A3 (ZnT3) oligomerization by dityrosine bonds regulates its subcellular localization and metal transport capacity. PloS one 4, e5896 (2009).
Salazar, G. et al. Phosphatidylinositol-4-kinase type II alpha is a component of adaptor protein-3-derived vesicles. Mol Biol Cell 16, 3692–3704 (2005).
Reid, C. A. et al. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology 80, 1003–1008 (2013).
Reid, C. A. et al. Reduced dendritic arborization and hyperexcitability of pyramidal neurons in a Scn1b-based model of Dravet syndrome. Brain 137, 1701–1715 (2014).
Richards, K. L. et al. Hippocampal volume and cell density changes in a mouse model of human genetic epilepsy. Neurology 80, 1240–1246 (2013).
Wimmer, V. C. et al. Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J Clin Invest 120, 2661–2671 (2010).
Scheffer, I. E. & Berkovic, S. F. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 120 (Pt 3), 479–490 (1997).
Escayg, A. et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS +2. Nat Genet 24, 343–345 (2000).
Schubert, J. et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 46, 1327–32 (2014).
Singh, N. A. et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 5, e1000649 (2009).
Wallace, R. H. et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 28, 49–52 (2001).
Wallace, R. H. et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+ -channel beta1 subunit gene SCN1B. Nat Genet 19, 366–370 (1998).
Feenstra, B. et al. Common variants associated with general and MMR vaccine-related febrile seizures. Nat Genet 46, 1274–82 (2014).
Hessel, E. V. et al. Identification of Srp9 as a febrile seizure susceptibility gene. Ann Clin Transl Neurol 1, 239–250 (2014).
Dibbens L. M. et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol 67, 542–546 (2010).
Nakamura, Y. et al. Novel HCN2 mutation contributes to febrile seizures by shifting the channel’s kinetics in a temperature-dependent manner. PloS One 8, e80376 (2013).
Puranam, R. S. et al. Disruption of Fgf13 Causes Synaptic Excitatory-Inhibitory Imbalance and Genetic Epilepsy and Febrile Seizures Plus. J Neurosci 35, 8866–8881 (2015).
Puskarjov, M. et al. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Reports 15, 723–729 (2014).
Paoletti, P., Vergnano, A. M., Barbour, B. & Casado, M. Zinc at glutamatergic synapses. Neuroscience 158, 126–136 (2009).
Smart, T. G., Hosie, A. M. & Miller, P. S. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist 10, 432–442 (2004).
Veran, J. et al. Zinc potentiates GluK3 glutamate receptor function by stabilizing the ligand binding domain dimer interface. Neuron 76, 565–578 (2012).
Chen, N., Moshaver, A. & Raymond, L. A. Differential sensitivity of recombinant N-methyl-D-aspartate receptor subtypes to zinc inhibition. Mol Pharmacol 51, 1015–1023 (1997).
Mizunuma, M., Takahashi, N., Usami, A., Matsuki, N. & Ikegaya, Y. High-temperature, but not high-pressure, conditions alter neuronal activity. J Pharmacol Sci 110, 117–121 (2009).
da Rocha, T. J. et al. SLC30A3 and SEP15 gene polymorphisms influence the serum concentrations of zinc and selenium in mature adults. Nutr Res 34, 742–748 (2014).
Wimmer, V. C. et al. Sodium channel beta1 subunit localizes to axon initial segments of excitatory and inhibitory neurons and shows regional heterogeneity in mouse brain. J Comp Neurol 523, 814–830 (2015).
Adlard, P. A., Parncutt, J. M., Finkelstein, D. I. & Bush, A. I. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J Neurosci 30, 1631–1636 (2010).
Acknowledgements
We thank the families for their participation in this study. Elena Aleksoska (Epilepsy Research Centre) is acknowledged for performing genomic DNA extractions. Technical expertise was provided by the Flow Cytometry Facility, MBC, Parkville and the Biometals Facility, MBC, Parkville. We thank Rosie Hartie for help with the imaging experiments. We acknowledge the contribution of the VIB Genetic Service Facility for the genetic follow-up analyses (http://www.vibgeneticservicefacility.be). The Florey Institute of Neuroscience and Mental Health is supported by Victorian State Government infrastructure funds. This work was supported by National Health and Medical Research Council (NHMRC) Program Grant (628952) to S.F.B., S.P. and I.E.S., an Australia Fellowship (466671) to S.F.B, a Practitioner Fellowship (1006110) to I.E.S, a Postdoctoral Training Fellowship to S.A.M and a Career Development Fellowship (1063799) to M.S.H. C.A.R is supported by a Dowd Fellowship. K.H. is a PhD fellow of the Institute for Science and Technology (IWT)-Flanders and A.S. is a postdoctoral fellow of the FWO.
Author information
Authors and Affiliations
Contributions
C.A.R. initiated the project. C.A.R., A.M.P., S.A.M. and M.S.H. directed the project. S.A.M., S.F.B., I.E.S., S.T.B., J.M.M., R.B., S.W. and P.D.J. conducted clinical phenotyping. M.S.H., J.A.D., K.H., R.H. and A.S. performed sequencing. C.A.R., A.M.P., V.W. and S.P. constructed mutant ZNT3, completed Zn2+ synaptic uptake, immunohistochemistry and imaging and tested thermogenic seizures in mouse models. P.A.A measured Zn2+ levels. C.A.R., A.M.P., S.A.M. and M.S.H. wrote the paper. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
I.E.S. discloses payments from UCB Pharma, Athena Diagnostics and Transgenomics for lectures and educational presentations. S.F.B. discloses payments from UCB Pharma, Novartis Pharmaceuticals, Sanofi-Aventis and Jansen Cilag for lectures and educational presentations and a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hildebrand, M., Phillips, A., Mullen, S. et al. Loss of synaptic Zn2+ transporter function increases risk of febrile seizures. Sci Rep 5, 17816 (2015). https://doi.org/10.1038/srep17816
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17816
This article is cited by
-
Serum Levels of Growth-Associated Protein-43 and Neurotrophin-3 in Childhood Epilepsy and Their Relation to Zinc Levels
Biological Trace Element Research (2023)
-
Zinc Supplementation for Prevention of Febrile Seizures Recurrences in Children: A Systematic Review and Meta-Analysis
Indian Pediatrics (2021)
-
Serum zinc and copper levels in a sample of Egyptian epileptic children
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2020)
-
Aberrant expression of PAR bZIP transcription factors is associated with epileptogenesis, focus on hepatic leukemia factor
Scientific Reports (2020)
-
Why and how to investigate the role of protein phosphorylation in ZIP and ZnT zinc transporter activity and regulation
Cellular and Molecular Life Sciences (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
