
Abstract
Bioprocess limitations imposed by microbial cell-to-cell phenotypic diversity remain poorly understood. To address this, we investigated the origins of such culture diversity during lipid production and assessed the impact of the fermentation microenvironment. We measured the single-cell lipid production dynamics in a time-invariant microfluidic environment and discovered that production is not monotonic, but rather sporadic with time. To characterize this, we introduce bioprocessing noise and identify its epigenetic origins. We linked such intracellular production fluctuations with cell-to-cell productivity diversity in culture. This unmasked the phenotypic diversity amplification by the culture microenvironment, a critical parameter in strain engineering as well as metabolic disease treatment.
Similar content being viewed by others

Introduction
The emerging paradigm of constructing target phenotypes for the production of chemical products and biofuels is attracting considerable interest and has met with significant success in recent years1,2,3,4. Despite substantial progress, however, engineered strains in culture do not typically reach the theoretical maximum, questioning thus the strain’s phenotypic robustness. Additionally, single cell analysis reveals a considerable productivity variance within a clonal population. Whereas few individual cells greatly outperform the median productivity, others lag in productivity and are even less efficient than non-engineered strains. This in essence limits our ability to register the true phenotype of a construct and, as such, it limits its bioprocessing reliability.
Here we investigate the aforementioned limitations in bioprocesses at the single cell level with a specific focus on the de-novo lipid biogenesis of Yarrowia lipolytica, a promising candidate in the production of oleochemicals5,6,7. First, we determined the phenotypic diversity in culture during batch growth of an over-producing and an under-producing strain. Subsequently, the lipid production dynamics of both strains were analyzed at the single cell level using microfluidics, primarily for two reasons. The first was to perform longitudinal investigations by tracking the lipid content at the single cell level over time. The second was to deterministically control the extracellular hydrodynamics, thus generating a quasi-time-invariance through ultra-fast nutrient supply and byproduct removal. Under such conditions, the inherent intracellular fluctuations of lipid abundance within an ideal extracellular environment were unmasked. We identified the epigenetic origins of such fluctuations and then compared them to the lipid content diversity between individuals in culture, similar to recent gene expression noise investigations8,9,10,11,12. This revealed that extracellular microheterogeneities during fermentation amplify intracellular fluctuations, thereby placing an upper limit in the culture’s productivity and bioprocessing reliability.
Results
Phenotypic Diversity in Culture: The Po1g and MTYL053 strains of Yarrowia lipolytica were investigated, as previously developed and extensively characterized5. Po1g and MTYL053 are identical except for leucine auxotrophy in Po1g and an intron-containing translation elongation factor-1α (TEF) promoter for over overexpressing diacylglycerol acyltransferase (DGA1) in MTYL053. As a result, the expression levels of DGA1 in MTYL053 are 17-fold enhanced compared to the intronless TEF promoter, as previously measured in detail5 (also qualitatively shown in Fig. S1). DGA1 is the final step of the triglyceride synthesis pathway thereby enabling MTYL053 with enhanced lipid production5. To evaluate the phenotypic diversity in culture, image cytometry13 was employed (Supplementary Information) to define the size and number/size of lipid droplets (LDs) in individual cells. As shown in Fig. 1a, the median lipid content (Si—normalized over the cell size) during early stationary phase (Fig. S2) was 4.1% for Po1g and – as expected – considerably higher for MTYL053 (14.2%).

(a) A scatter plot illustrating the neutral lipid content normalized over the cell area for individual Po1g (red) and MTYL053 (blue) cells; the median lipid content for each strain is represented with a solid line. (b) The frequency distribution of the lipid content per cell for the two strains, (c) Microfluidic immobilization of single Po1g cells under continuous laminar flow at a 1 μL/min rate (top). In the lower snapshot series at 20′ time-steps, composite images of the cell (bright-field) and its neutral lipid load (green) illustrate the fluctuation of the cell’s lipid content (Si). (d) Longitudinal traces of the lipid content per cell for Po1g (upper) and MTYL053 (lower); the lineage of an individual cell is highlighted in blue. (e) Boxcharts (range: 25–75%) illustrating the single-cell bioprocessing noise for Po1g (upper) and MTYL053 (lower); the whisker lines denote the outliers.
Despite their different productivities, both strains suffered from a considerable lipid content diversity. This is best depicted by the distributions in (Fig. 1b), denoting the co-existence of individuals with lipid content lower or higher than the median for both strains. The phenotypic diversity was determined through the robust coefficient of variation (rCV—Supplementary Information), defined as the ratio of the robust standard deviation over the median. The rCV was employed to define the deviation of individual cells from the population’s median, instead of conventional estimates of central tendencies (mean) and dispersion (standard deviation). The latter assume the data form a random sample from a normal distribution. This is not the case in our experiments due to the presence of outliers as typically observed in single-cell analyses, such as the recent lipid content heterogeneity investigations using flow cytometry14. Under the aforementioned conditions, the rCV is similar to phenotypic robustness and thus linked to strain reliability (R) through the relationship: R = 1 – rCV15,16. The reliability factors for Po1g was 0.31 and moderately improved for MTYL053 (0.5). Indicative of the similar reliability factors is the considerable phenotypic overlap of MTYL053 with the non-engineered Po1g (Fig. 1b). This questions our ability to register the true phenotype of the engineered strain, as well as the reliability of the pathway engineering strategy17.
Single-Cell Lipid Production Dynamics: To investigate this phenotypic overlap, we performed a time-dependent analysis of lipid production at the single cell level. Both single Po1g and MTYL053 cells were immobilized using microfluidics (Fig. 1c) and their lipid content was measured every 20 min using confocal microscopy and vesicle photonics18 (Supplementary Information). The protein content was not simultaneously monitored in this instance, in order to avoid perturbations to cell physiology (e.g. cell lysis for single-cell qRT-PCR), or temporal resolution limitations in wavelength-multiplexed confocal imaging due to the high variability in maturation times of fluorescent proteins19. During imaging, laminar microflows (1 μL/min rates) were applied, enabling rapid replenishment of the extracellular environment at frequencies greater than 10 cell volumes/msec. This generated a quasi-time invariant extracellular microenvironment, a critical aspect in our study given the rapid metabolome dynamics with perturbation response times as low as 1 sec or less20. The employed flow rates also applied mechanical forces on the cells; however, these are in the 10 pN range, therefore exerting insignificant mechanical stress given the enhanced surface modulus of yeast21. Prior to microfluidic immobilization, cells were harvested at early stationary phase, thus enabling direct comparison with the aforementioned phenotypic diversity analysis, as well as prolonged viability, enhanced lipid content and low budding probability (Supplementary Information and Fig. S3, S4).
Under such conditions, lipid accumulation in either strain was not monotonic, but rather fluctuated with time through continuous cycles of replication and degradation (Fig. 1d). This unmasks an inherent form of noise in biosynthesis, termed bioprocessing noise (bp). Similar forms of noise have been previously defined in gene expression8,22,23,24,25,26,27,28 and recently in cell growth29 and metabolic pathway use30. We attribute the observed noise during biosynthesis to the sporadic nature of both gene expression and metabolic reactions. In essence both types of reactions exhibit an efficiency that depends on the abundance, activity and locations of all involved reactants, namely regulatory elements during gene expression, as well as metabolites, enzymes and co-factors during metabolic synthesis or degradation. Such copy-number and state fluctuations give rise in turn to a probability distribution of the reaction product itself. Similar to the nomenclature of gene expression noise8,22,23,24,25,26,27,28, the ‘extrinsic’ and ‘intrinsic’ contributions to bioprocessing noise emanate from variability in enzyme production and variability at the metabolic reaction level respectively. The single-cell bioprocessing noise was quantified through the robust coefficient of variation of the lipid content longitudinal trace (Fig. 1d). The median noise for Po1g was bpPo1g = 0.35, while the transformed strain MTYL053 exhibited substantially less noise by approximately 6-fold (bpMTYL053 = 0.06) (Fig. 1e).
Discussion
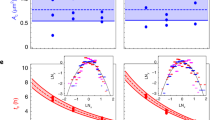
It becomes apparent that enzyme over-expression in MTYL053 not only induces enhanced lipid production but also reduces bioprocessing noise. This is better visualized in Fig. 2a that plots the median lipid content and bioprocessing noise between the two strains (cross points). An analogous dependence also persists within individual cells in each strain (Fig. 2a – scatter plot), with bioprocessing noise scaling inversely with the square root of lipid content in both strains. Interestingly, bioprocessing noise was independent of cell size for both strains, eliminating thus growth rate or stage variations as possible sources of the observed noise differences between individuals (Fig. 2a-inset).

(a) Bioprocessing noise as a function of the inverse square root of lipid content; each data point denotes the median per cell for Po1g (red) and MTYL053 (blue) with the larger cross-points indicating the median per strain interconnected through a linear fit. Inset histogram plots the correlation coefficients between bioprocessing noise with cell size for both Po1g and MTYL053. (b) The non-linear dependence of population level reliability (R) and phenotypic diversity on bioprocessing noise (bp) for Po1g, Po1g + CHX and MTYL053; the red line represents the exponential decay fit of the form: R = Ro + A∙eα∙bp, where Ro = 0.5, A = −0.003 and α = 12. Inset compares the bioprocessing noise box-charts for Po1g and Po1g + CHX.
It is relevant to compare the aforementioned inverse scaling with a similar one frequently encountered in gene expression noise8,22. Therein, gene expression noise also scales inversely with the square root of protein abundance, especially for proteins of low and medium abundances. This is associated with the spontaneous rate fluctuations in gene expression, emanating from micro- heterogeneities and crowding of the cytosol and cell-cycle dependent protein copy numbers8. Our interpretation of the above in bioprocesses is that in addition to gene expression, metabolic reactions are also subject to spontaneous rate fluctuations. Consequently, a continuous probability distribution within individual cells for both enzyme and metabolite copy numbers is enforced, contributing thus to the overall bioprocessing noise.
To compare the bioprocessing noise contributions of gene expression and metabolic reaction fluctuations, we added cycloheximide (CHX) to the YPD media (33 μg/ml) and repeated the dynamic analysis on the noisiest strain (Po1g). CHX terminates eukaryotic protein synthesis (and inhibits growth Fig. S2), thus diminishing noise contributions during transcription or translation31. Indeed, the addition of CHX reduced bioprocessing noise by approximately 15% in Po1g (Fig. 2b – inset). This evidences that gene expression fluctuations do contribute to bioprocessing noise. Bioprocessing noise however is not completely diminished under CHX admission, demonstrating that contributions from metabolic reaction fluctuations are also present and likely more dominant. It is worth noting that the randomness of gene expression and metabolic reactions are known to follow an epigenetic/non-Mendelian heredity pattern8. This indicates that bioprocessing noise is an intrinsic characteristic of a culture, bounded largely by enzyme expression levels as evidenced by the Po1g—MTYL053 comparison.
The question however persists: if bioprocessing noise is lower in MTYL053 than Po1g in microfluidics, then why do the two strains exhibit comparable reliability in culture during batch growth? To answer this, we plotted the reliability factor (R) of all three cases (Po1g, Po1g-CHX and MTYL053) against bioprocessing noise (bp) in the same graph (Fig. 2b). The plot indicates that ‘R’ and ‘bp’ are inversely related, namely noisier strains are also less reliable and exhibit higher phenotypic diversity (right y-axis). The plot reveals two additional characteristics. The first is that phenotypic diversity is persistently higher (>0.5) than bioprocessing noise (<0.5) for all cases. We attribute this to the different extracellular microenvironment between batch growth and microfluidics. During fermentation, the extracellular time invariance in microfluidics is replaced by limited mixing that induces microheterogeneities such as chemical gradients of key nutrients or waste products. These amplify intracellular bioprocessing noise during fermentation, enhancing population diversity in culture. Specifically for Yarrowia lipolytica at early exponential phase (Fig. 2b), the upper reliability (or diversity) limit set by the fermentation microenvironment was Ro ~ 0.5 at zero-amplitude intracellular fluctuations (bp = 0). The second feature is the non-linear dependence of reliability on bioprocessing noise as indicated by the exponential decay curve fitting in Fig. 2b. This nonlinear amplification of intracellular noise is likely due to low autoregulation between extracellular events with intracellular reactions, in contrast to the established feedback stabilizing mechanisms in gene networks32.
In conclusion, we employed microfluidics for comparing bioprocessing dynamics at the single-cell level in a controlled microenvironment with cell-to-cell bioprocessing diversity in culture during batch growth. Within a time-invariant microfluidic environment, we observed that lipogenesis is sporadic with time, rather than monotonic. Such an observation can only be achieved through the longitudinal analysis of single cells, otherwise impossible at the culture level, or through alternative single cell methods such as flow cytometry. In light of this, we introduced bioprocessing noise and identified its epigenetic origins. Subsequently, we compared the time-dependent single cell behavior in microfluidics with the persistent cell-to-cell variability during batch growth. The use of microfluidics was also critical here, by enabling deterministic control of the extracellular environment. Through this, we discovered that intracellular lipid production fluctuations are non-linearly linked with extracellular events, a critical insight to biofuels production, as well as medical implications of fatty acids, such as in liver disease14, adipogenesis33 and cancer34. Microfluidics has been crucial in screening gene and protein interactions and resulting complex phenotypes at the single cell level35,36. Our results expand the possibilities of such microsystems to identifying the true phenotype of a strain, which is otherwise masked by microheterogeneities within the extracellular environment. Additionally, such microsystems enable the accurate control or assessment of the extracellular microenvironment on the efficiency of metabolic processes, thus enabling enhanced bioprocess investigations and potentially optimized bioreactor designs.
Materials and Methods
The experiments were performed using the two derivatives Po1g and MTYL053 of the oleaginous yeast Yarrowia lipolytica. More detailed information on the auxotrophic wild type strain (Po1g – Leu-) and the engineered one (MTYL053) are included in reference5. The cells were grown at room temperature (regulated at 25 °C ± 1 °C) until early stationary phase in YPD media, with glucose as the carbon source (10 g/L Yeast Extract – Difco Laboratories, 20 g/L Bacto-Peptone – Difco and 20 g/L glucose – Sigma Aldrich). To visualize the lipid droplets, the bodipy dye (BODIPY® 493/503 (4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene – Molecular Probes) solution in DMSO (Molecular Probes) was added in YPD media at a concentration of 250 ng/ml, followed by a 4 hour long incubation under the same temperature and shaking conditions as those maintained during batch growth. Specific to the microfluidic experiments, following the 4 hour long staining period, the cells were transferred to a gastight syringe at a 10x dilution in YPD media containing a lower concentration of the bodipy dye (100 ng/ml) and 0.02% DMSO and loaded to the microfluidics through one inlet (Fig. S5a). Following immobilization, the same solution (YPD-Bodipy concentration 100 ng/ml, 0.02% DMSO) was continuously supplied through a second inlet at a rate of 1 μL/min to enable stable immobilization. On average ~n = 90 individual cells per strain and growth condition were studied.
The microfluidics were fabricated with conventional soft lithography by molding polydimethilsiloxane (PDMS, Sylgrad 184, Dow Corning) and bonding on a glass coverslip37. Briefly, a 10:1 monomer to catalyst ratio was manually mixed and degassed for 2 hour in a vacuum desiccator. The mixture was subsequently poured over a mask (SU8 photoresist on a silicon substrate) and baked for 2 hours at 70 °C, followed by cutting using a razor blade, plasma assisted bonding to glass coverslips and overnight baking at 70 °C. Two inlets were integrated, one for cell loading and one for media delivery; one outlet enabled waste collection (Fig. S5a). Media and cell flow were enabled by two syringe pumps (Harvard Apparatus) and Tygon tubing (inner diameter 0.02″).
The cell trapping design was based on the Tan and Takeuchi axial percolation filtration method and the principle of path of minimal hydrodynamic resistance38; briefly, 16 trapping sites were arrayed connected to a cell-loading and a media-delivering channel (50 μm wide – 10 μm thick). The traps comprised of a dwell chamber and a narrow indentation with approximate widths 6 μm and 2.5 μm respectively (Fig. S5b)39. Given the channel’s height being almost double than the cell’s, we anticipate that fluid flow also occurs through the narrow indentation even when it is infilled with an individual cell. On average, 6–8 cells were trapped per experiment, resulting in an approximately 50% trapping efficiency; however, such a reduced trapping efficiency did not limit our experimental throughput, due to temporal limitations placed by optical sampling. The latter necessitated approximately 1–1.5 min dwell time to complete the confocal imaging at three wavelength channels, namely green excitation for lipid detection, red excitation for viability detection and brightfield for cell size determination. Such dwell times imposed the upper limit to the temporal resolution of the lipid expression fluctuation analysis, which we accommodated by selecting a time step of 20 min for the whole cell array (more information is included in the Supplementary Information). The cells at late stationary phase remained viable for more than 9 hours. This was determined by adding the propidium iodide dye (Sigma Aldrich) in the media (Fig. S3). Confocal fluorescent imaging was employed to determine the lipid content in individual cells. This imaging modality, contrary to higher resolution and specificity approaches, such as Electron Microscopy, enabled long term longitudinal imaging of individual cells under minimal perturbations to cell physiology. Further information on cell growth, staining, imaging and data analysis, as well as supplementary images are available within the online Supplementary Information.
Additional Information
How to cite this article: Vasdekis, A. E. et al. Origins of Cell-to-Cell Bioprocessing Diversity and Implications of the Extracellular Environment Revealed at the Single-Cell Level. Sci. Rep. 5, 17689; doi: 10.1038/srep17689 (2015).
References
Stephanopoulos, G. Challenges in engineering microbes for biofuels production. Science 315, 801–804, doi: 10.1126/science.1139612 (2007).
Zhang, F., Carothers, J. M. & Keasling, J. D. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nature Biotechnology 30, 354–U166, doi: 10.1038/nbt.2149 (2012).
Steen, E. J. et al. Microbial production of fatty-acid-derived .fuels and chemicals from plant biomass. Nature 463, 559–U182, doi: 10.1038/nature08721 (2010).
Atsumi, S., Hanai, T. & Liao, J. C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451, 86–U13, doi: 10.1038/nature06450 (2008).
Tai, M. & Stephanopoulos, G. Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metabolic Engineering 15, 1–9, doi: 10.1016/j.ymben.2012.08.007 (2013).
Blazeck, J. et al. Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nature Communications 5, doi: 10.1038/ncomms4131 (2014).
Beopoulos, A. et al. Control of Lipid Accumulation in the Yeast Yarrowia lipolytica. Applied and Environmental Microbiology 74, 7779–7789, doi: 10.1128/aem.01412-08 (2008).
Kaern, M., Elston, T. C., Blake, W. J. & Collins, J. J. Stochasticity in gene expression: From theories to phenotypes. Nature Reviews Genetics 6, 451–464, doi: 10.1038/nrg1615 (2005).
Acar, M., Mettetal, J. T. & van Oudenaarden, A. Stochastic switching as a survival strategy in fluctuating environments. Nature Genetics 40, 471–475, doi: 10.1038/ng.110 (2008).
Balazsi, G., van Oudenaarden, A. & Collins, J. J. Cellular Decision Making and Biological Noise: From Microbes to Mammals. Cell 144, 910–925, doi: 10.1016/j.cell.2011.01.030 (2011).
So, L.-H. et al. General properties of transcriptional time series in Escherichia coli. Nature Genetics 43, 554–U584, doi: 10.1038/ng.821 (2011).
Sorek, M., Balaban, N. Q. & Loewenstein, Y. Stochasticity, Bistability and the Wisdom of Crowds: A Model for Associative Learning in Genetic Regulatory Networks. Plos Computational Biology 9, doi: 10.1371/journal.pcbi.1003179 (2013).
Vasdekis, A. E. & Stephanopoulos, G. Review of methods to probe single cell metabolism and bioenergetics. Metabolic Engineering 27, 115–135, doi: 10.1016/j.ymben.2014.09.007 (2015).
Herms, A. et al. Cell-to-Cell Heterogeneity in Lipid Droplets Suggests a Mechanism to Reduce Lipotoxicity. Current Biology 23, 1489–1496, doi: 10.1016/j.cub.2013.06.032 (2013).
Shechtman, O. The Coefficient of Variation as an Index of Measurement Reliability. Vol. 3, 39–49 (Springer, 2013).
Yang, L., Srinivasan, S., Mahadevan, R. & Cluett, W. R. Characterizing metabolic pathway diversification in the context of perturbation size. Metabolic Engineering 28, 114–122, doi: 10.1016/j.ymben.2014.11.013 (2015).
Klein-Marcuschamer, D. & Stephanopoulos, G. Assessing the potential of mutational strategies to elicit new phenotypes in industrial strains. Proceedings of the National Academy of Sciences of the United States of America 105, 2319–2324, doi: 10.1073/pnas.0712177105 (2008).
Vasdekis, A. E., Scott, E. A., Roke, S., Hubbell, J. A. & Psaltis, D. Vesicle Photonics. Annual Review of Materials Research, Vol 43 43, 283–305, doi: 10.1146/annurev-matsci-071312-121724 (2013).
Hebisch, E., Knebel, J., Landsberg, J., Frey, E. & Leisner, M. High Variation of Fluorescence Protein Maturation Times in Closely Related Escherichia coli Strains. Plos One 8, doi: 10.1371/journal.pone.0075991 (2013).
Zenobi, R. Single-Cell Metabolomics: Analytical and Biological Perspectives. Science 342, 1201-+, doi: 10.1126/science.1243259 (2013).
Smith, A. E., Zhang, Z. B., Thomas, C. R., Moxham, K. E. & Middelberg, A. P. J. The mechanical properties of Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 97, 9871–9874, doi: 10.1073/pnas.97.18.9871 (2000).
Elowitz, M. B., Levine, A. J., Siggia, E. D. & Swain, P. S. Stochastic gene expression in a single cell. Science 297, 1183–1186, doi: 10.1126/science.1070919 (2002).
Ozbudak, E. M., Thattai, M., Kurtser, I., Grossman, A. D. & van Oudenaarden, A. Regulation of noise in the expression of a single gene. Nature Genetics 31, 69–73, doi: 10.1038/ng869 (2002).
Swain, P. S., Elowitz, M. B. & Siggia, E. D. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proceedings of the National Academy of Sciences of the United States of America 99, 12795–12800, doi: 10.1073/pnas.162041399 (2002).
Mettetal, J. T., Muzzey, D., Pedraza, J. M., Ozbudak, E. M. & van Oudenaarden, A. Predicting stochastic gene expression dynamics in single cells. Proceedings of the National Academy of Sciences of the United States of America 103, 7304–7309, doi: 10.1073/pnas.0509874103 (2006).
Raj, A. & van Oudenaarden, A. Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 135, 216–226, doi: 10.1016/j.cell.2008.09.050 (2008).
Munsky, B., Neuert, G. & van Oudenaarden, A. Using Gene Expression Noise to Understand Gene Regulation. Science 336, 183–187, doi: 10.1126/science.1216379 (2012).
Li, G.-W. & Xie, X. S. Central dogma at the single-molecule level in living cells. Nature 475, 308–315, doi: 10.1038/nature10315 (2011).
Kiviet, D. J. et al. Stochasticity of metabolism and growth at the single-cell level. Nature 514, 376-+, doi: 10.1038/nature13582 (2014).
Labhsetwar, P., Cole, J. A., Roberts, E., Price, N. D. & Luthey-Schulten, Z. A. Heterogeneity in protein expression induces metabolic variability in a modeled Escherichia coli population. Proceedings of the National Academy of Sciences of the United States of America 110, 14006–14011, doi: 10.1073/pnas.1222569110 (2013).
Belle, A., Tanay, A., Bitincka, L., Shamir, R. & O’Shea, E. K. Quantification of protein half-lives in the budding yeast proteome. Proceedings of the National Academy of Sciences of the United States of America 103, 13004–13009, doi: 10.1073/pnas.0605420103 (2006).
Becskei, A. & Serrano, S. Engineering stability in gene networks by autoregulation. Nature 405, 590–593 (2000).
Loo, L.-H. et al. Heterogeneity in the physiological states and pharmacological responses of differentiating 3T3-L1 preadipocytes. Journal of Cell Biology 187, 375–384, doi: 10.1083/jcb.200904140 (2009).
Gocze, P. M. & Freeman, D. A. Factors Underlying the Variability of Lipid Droplet Fluorescence in MA-10 Leydig Tumor Cells. Cytometry 17, 151–158, doi: 10.1002/cyto.990170207 (1994).
Tay, S. et al. Single-cell NF-kappa B dynamics reveal digital activation and analogue information processing. Nature 466, 267–U149, doi: 10.1038/nature09145 (2010).
Wang, B. L. et al. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nature Biotechnology 32, 473–U194, doi: 10.1038/nbt.2857 (2014).
Thorsen, T., Maerkl, S. J. & Quake, S. R. Microfluidic large-scale integration. Science 298, 580–584, doi: 10.1126/science.1076996 (2002).
Tan, W.-H. & Takeuchi, S. A trap-and-release integrated microfluidic system for dynamic microarray applications. Proceedings of the National Academy of Sciences of the United States of America 104, 1146–1151, doi: 10.1073/pnas.0606625104 (2007).
Vasdekis, A. E. Single microbe trap and release in sub-microfluidics. RSC Advances 3, 6343–6346, doi: 10.1039/c3ra40369f (2013).
Acknowledgements
GS acknowledges financial support from the US Department of Energy, Grant no. SC 0008744 and AEV from an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20 GM103408), as well as from the Pacific Northwest National Laboratory (Linus Pauling Fellowship – PN12005/2406). Part of the research was performed using EMSL, a national user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research located at Pacific Northwest National Laboratory.
Author information
Authors and Affiliations
Contributions
A.E.V. and G.S. designed the research and wrote the manuscript; A.E.V. performed the experiments and data analysis; A.M.S. prepared the cell samples and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vasdekis, A., Silverman, A. & Stephanopoulos, G. Origins of Cell-to-Cell Bioprocessing Diversity and Implications of the Extracellular Environment Revealed at the Single-Cell Level. Sci Rep 5, 17689 (2016). https://doi.org/10.1038/srep17689
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17689
This article is cited by
-
Eliciting the impacts of cellular noise on metabolic trade-offs by quantitative mass imaging
Nature Communications (2019)
-
Synthetic biology for manufacturing chemicals: constraints drive the use of non-conventional microbial platforms
Applied Microbiology and Biotechnology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
