
Abstract
Live cell RNA imaging using genetically encoded fluorescent labels is an important tool for monitoring RNA activities. A recently reported RNA aptamer-fluorogen system, the Spinach, in which an RNA aptamer binds and induces the fluorescence of a GFP-like 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI) ligand, can be readily tagged to the RNA of interest. Although the aptamer–fluorogen system is sufficient for imaging highly abundant non-coding RNAs (tRNAs, rRNAs, etc.), it performs poorly for mRNA imaging due to low brightness. In addition, whether the aptamer-fluorogen system may perturb the native RNA characteristics has not been systematically characterized at the levels of RNA transcription, translation and degradation. To increase the brightness of these aptamer-fluorogen systems, we constructed and tested tandem arrays containing multiple Spinach aptamers (8–64 aptamer repeats). Such arrays enhanced the brightness of the tagged mRNA molecules by up to ~17 fold in living cells. Strong laser excitation with pulsed illumination further increased the imaging sensitivity of Spinach array-tagged RNAs. Moreover, transcriptional fusion to the Spinach array did not affect mRNA transcription, translation or degradation, indicating that aptamer arrays might be a generalizable labeling method for high-performance and low-perturbation live cell RNA imaging.
Similar content being viewed by others


Introduction
RNAs play diverse functions in living cells, including delivering genetic information, catalyzing chemical reactions and regulating gene expression at multiple levels1,2. Recent genome-wide analysis has suggested that inhomogeneous RNA localization within different cellular compartments might be more prevalent than previously appreciated3,4, resulting in highly localized spatio-temporal modulations in gene expression levels within those subcellular compartments. Compared to biochemical approaches, such as northern blot, quantitative reverse transcription PCR (qPCR), high-throughput RNA sequencing, etc., direct visualization of RNAs by fluorescence imaging allows spatial and temporal RNA tracking and is capable of correlating transcription, localization, translation and degradation of RNAs and simultaneously revealing cell-to-cell heterogeneity5,6,7. Nevertheless, unlike imaging proteins, there are not many tools for imaging RNAs. Fluorescence in situ hybridization (FISH) utilizes fluorescent dye-conjugated oligonucleotides (fluorescent probes) complementary to target RNAs to directly label RNA molecules and has been widely applied to accurately quantify the expression level and to localize distribution of mRNAs in biological samples8,9. Introduction of the fluorescence probes usually requires cell fixation and permeabilization fixed cell8. Alternative delivery methods compatible with live cell imaging include microinjection10,11, electroporation3, or transfection using polycationic molecules such as liposomes and dendrimers12 and membrane permeabilization via cell-penetrating peptides13 and streptolysin O14,15. However, these methods sometimes lead to problems such as cell damage, inhomogeneous probe delivery and inefficient probe annealing to target RNAs12.
The second type of RNA imaging utilizes indirect labeling and employs fluorescent fusion proteins and specific protein-RNA interactions, such as the RNA bacteriophage MS2 coat protein system16,17,18, the PP7 bacteriophage system19,20, the bacteriophage λ N coat protein system21, etc.22,23. In those strategies, RNAs of interest are tagged with a “cognate” RNA sequence, often in a tandem array, recognized and bound by interacting proteins with fluorescent protein fusion. There are several potential limitations associated with this type of methods: (1) usually the overexpressed unbound proteins generate high fluorescent background24, unless specific measures are taken to reduce the background22,23,25; (2) the resulting large ribonucleoprotein complex has been reported to affect the RNA endogenous degradation in some cases26,27 and may potentially affect RNA trafficking and localization11; (3) the conditions required for maturation of the fluorescent proteins prohibit the application of these approaches to certain biological systems, such as anaerobic species28,29,30.
More direct ways for RNA labeling in living cells mostly use RNA aptamers that can bind small ligands (or “fluorogens”) and activate their fluorescence31,32,33. Among several aptamer-ligand combinations, “Spinach” and analogous systems (Spinach34, Spinach 235, RNA Mango36, Broccoli37, etc.) have shown the greatest potential in biochemical assay and live cell imaging34,35,36,37,38,39,40,41,42,43,44,45. Spinach uses a short RNA aptamer (24-2 and 24-2-min RNA sequences, reported by Paige and coworkers34; ~100 nucleotides) that exhibits EGFP-like green fluorescence upon binding of 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), a fluorogenic ligand that is structurally similar to the EGFP chromophore and is membrane-permeable and nontoxic. Spinch RNA folds into an RNA G-quadruplex structure providing a binding site for the fluorogen46,47,48 and structural stabilization of the fluorogen bound to the G-quadruplex structure is likely responsible for its strong fluorescence enhancement. Although the Spinach system has been used for imaging highly abundant nontranslated RNAs (tRNAs34, rRNAs34,35,37 and trinucleotide repeats35) and further applied to detect cellular metabolites and proteins34,35,40,41,44, there have been very few studies on the utility of Spinach in imaging cellular mRNA42 and the fluorescence signal was only barely above cellular autofluorescence level, likely due to low abundance of mRNA and the low brightness of a single fluorogen-bound RNA aptamer. To amplify the signal, making a tandem array of these RNA aptamers, as was used for MS2 and PP7 systems26,27, could be a general solution to improve the brightness of the aptamer-fluorogen systems. However, it remains to be tested whether this strategy is suitable for RNA imaging because making tandem arrays of these RNA aptamers would significantly increase the RNA length which may potentially affect mRNA metabolism.
Here we use Spinach as our model system and fused multiple repeats of the Spinach aptamer in tandem on a single RNA molecule. We applied the tandem Spinach arrays to cellular mRNA imaging. In both in vitro measurement and cellular imaging, the Spinach array brought about as high as 17-fold fluorescence enhancement compared to single Spinach aptamer on RNAs, allowing us to image mRNA distributions inside living cells that could have not been achieved with single aptamer tagging. We also characterized the effects of such tandem arrays on mRNA transcription, translation, localization and degradation and found that the Spinach array does not alter these RNA characteristics, indicating that making tandem repeats of these aptamers might be a generally applicable strategy for mRNA live cell imaging.
Results
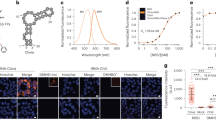
We designed a series of Spinach arrays containing different numbers of tandem Spinach aptamers (Spi-nR, n = 8, 16, 32, 64). Between two adjacent aptamer repeats we inserted 17-nt randomized spacer sequence, which was used for constructing tandem repeats of MS2 RNA sequence26,27. The Spinach array was inserted into pET28a plasmid, placing both in vitro and cellular RNA expression under the control of T7 promoter and lac operator system, respectively49 (Fig. 1A). Fluorescence intensities of in vitro transcribed Spinach arrays (100 nM RNA) were measured using a fluorometer after RNA folding and incubation with DFHBI (20 μM). Compared to single Spinach aptamer (Spi), Spi-nR shows greatly enhanced fluorescence signal with the same maximum excitation/emission wavelengths at 460/505 nm (Fig. 1B). The fluorescence intensity increases with the repeat number: it changes approximately 16-fold when the aptamer repeat number increases 64-fold (Fig. 1C), with the average 1.6-fold fluorescence enhancement upon repeat number duplication (Supplementary Methods and Materials and Supplementary Fig. S1). Binding of DFHBI to aptamers within the array showed similar kinetics (i.e, kon, koff and KD) as binding to single Spinach aptamer (Spi) (Supplementary Table S2).

(A) Sketch of in vitro transcription system for Spinach arrays (Spi-nR). (B) Excitation and emission spectra of single Spinach aptamer (Spi) and Spinach arrays (100 nM RNA +20 μM DFHBI). (C) Fluorescence intensities of Spi and Spi-nR, measured by fluorometer.
There are two possible explanations for the less than 2-fold increase in the brightness when the repeat number was doubled: fluorescence quenching between adjacent aptamers and misfolding/incomplete folding of multiple aptamers27,31. To test inter-aptamer quenching, we measured the fluorescence lifetimes of single Spinach and Spinach arrays. The average fluorescence lifetime of Spi, Spi-8R and Spi-32R was 4.0, 3.91 and 3.63 ns, respectively. Therefore, we estimated that quenching can account for only up to 10% in fluorescence reduction (Supplementary Methods and Materials). To test misfolding/incomplete folding, we used a previously reported assay to estimate the folding efficiency35,50 and found that the folding efficiency of Spi-8R relative to Spi is 43.5 ± 1.6% and drops further to 34.6 ± 2.0% for Spi-32R (Supplementary Table S1). Spinach is known to be subject to misfolding50 and Spi-nR may misfold more extensively through inter-aptamer misfolding, similar to the example where repeat proteins are known to misfold due to interdomain interactions51,52,53. Incorporating the single Spinach aptamer into a tRNA scaffold could slightly increase its fluorescence likely through improving the folding of the Spinach sequence (Supplementary Table S1 and Supplementary Fig. S2). However, recent study showed that the RNA aptamer incorporated into tRNA scaffold suffered endonucleolytic cleavage due to tRNA sequence recognition by RNases in bacteria and mammalian cells54, disfavoring the extensive use of tRNA scaffold in Spinach imaging.
In order to measure the fluorescence enhancement of Spinach array-tagged mRNA in living cells and characterize its influence on mRNA transcription, translation and degradation, we inserted Spinach arrays in the 3′ UTR (untranslated region) of the monomeric red fluorescent protein (mRFP1) coding sequence (henceforth called RFP-Spi-nR (n = 8, 16, 32, 64)) (Fig. 2A). For comparison, we also prepared two constructs each with a single Spinach: RFP-Spi, in which Spinach aptamer was directly linked to RFP sequence and RFP-Spi-tRNA, in which Spinach aptamer was incorporated into a human tRNALys scaffold as previously reported34,55. Quantitative reverse transcription PCR (qPCR) data showed that the cellular level of Spinach-tagged mRNA under induction (1 mM IPTG, 60 min), either by a single Spinach or by an array, is similar to that of untagged mRNA (Fig. 2B), suggesting that mRNA transcription is unperturbed by the Spinach tag even with the longest array tested. In addition, the average fluorescence intensities of mRFP1 protein per cell, translated from Spinach-tagged or untagged mRNAs, were comparable (Fig. 2C,D), indicating that mRNA translation is not affected by Spinach tag either as a single aptamer or as an array.

(A) Sketch of RFP-Spi-nR expression system in E. coli. (B) mRNA expression level of RFP or RFP-Spi-nR in E. coli after 60 min of 1 mM IPTG induction, measured by qPCR and normalized by the mRNA level of unmodified RFP in E. coli. (C) mRNA (Spinach) and protein (mRFP1) fluorescence in E. coli expressing RFP or RFP-Spi-nR upon induction, measured via epifluorescence imaging. (D) Representative fluorescence images of E. coli expressing RFP or RFP-Spi-nR upon induction.
We then measured how much brighter Spinach arrays are compared to single Spinach in living cells. Live cell imaging of E. coli transcribing RFP-Spi showed no fluorescence enhancement in the Spinach channel over the background autofluorescence level of cells expressing untagged RFP (Supplementary Fig. S3). In comparison, cells transcribing RFP-Spi-tRNA showed homogenously distributed Spinach fluorescence with a slight enhancement over the autofluorescence level (Fig. 2C,D and Supplementary Fig. S3). The fluorescence deficiency in cells transcribing RFP-Spi probably results from misfolding of the Spinach aptamer when fused to an mRNA, in line with the observation that in vitro transcribed and folded RFP-Spi did not show any Spinach fluorescence (Supplementary Fig. S2). In contrast, E. coli transcribing RFP-Spi-nR showed strong Spinach fluorescence. The increase in average fluorescence intensity per cell as a function of n (~17 fold enhancement from RFP-Spi-tRNA to RFP-Spi-64R, after autofluorescence subtraction) generally reflects the in vitro trend (~16 fold enhancement from Spi to Spi-64R). Moreover, distinct from the homogenously distributed fluorescence observed for RFP-Spi-tRNA, the fluorescence signal of RFP-Spi-nR preferentially accumulated at cell poles. In order to determine whether the apparent difference in cellular localization between the two constructs is caused by Spinach arrays, we conducted an RNA FISH experiment on cells expressing RFP, RFP-Spi-tRNA or RFP-Spi-nR (n = 8, 32), using Cy5-labeled oligonucleotide probes targeting the mRFP1 sequence (Supplementary Fig. S4) as well as the Spinach array (Supplementary Fig. S5). FISH data indicated that upon IPTG induction, mRNAs were preferentially accumulated at the cell poles where mRNAs transcribed from plasmid DNA are typically observed in E. coli56, regardless of the presence or the types of the Spinach tag at the 3′ UTR (Supplementary Fig. S4). Therefore, we conclude that the low fluorescence from single Spinach failed to reveal accurate mRNA localization (Fig. 2 and Supplementary Fig. S6) and the Spinach array could address the problem by greatly enhancing fluorescence signal for cellular mRNA imaging.
Next, we examined the effect of Spinach arrays on RNA degradation. We conducted an mRNA decay assay for E. coli cells expressing RFP-Spi-32R or untagged RFP. We monitored the fluorescence of RFP-Spi-32R mRNA and mRFP1 protein after IPTG removal which would stop the synthesis of target RNAs. Spinach fluorescence levels greatly decreased within 30 min and were depleted within 90 min after withdrawal of IPTG suggesting mRNA degradation (Fig. 3A,B). We also compared mRFP1 fluorescence between E. coli expressing RFP-Spi-32R and untagged RFP (Fig. 3A and Supplementary Fig. S7). In both cases the fluorescence drop showed a significant time lag behind Spinach fluorescence change, which possibly resulted from the combination of the following: (1) continuous translation of mRFP1 from the remaining mRNA after IPTG removal, (2) much longer lifetime of protein compared to that of mRNA57,58,59, and/or (3) mRFP1 maturation60,61. We observed very similar trends of mRFP1 fluorescence intensity change in cells expressing RFP-Spi-32R and RFP (Supplementary Fig. S7), indicating that the Spinach array does not affect mRNA degradation or translation kinetics. To further confirm the effect of Spinach array on mRNA degradation, we conducted qPCR experiment to measure the abundance of untagged RFP mRNA and RFP-Spi-nR (n = 8, 32) as a function of time after IPTG removal (Fig. 3C). qPCR data showed that RFP-Spi-nR mRNA level was decreased by approximately 60% and 90% 30 and 120 min after IPTG removal, respectively, with the same trend observed for the untagged RFP. We therefore conclude that Spinach arrays do not influence mRNA decay.

(A) mRNA (Spinach) and protein (mRFP1) fluorescence of E. coli expressing RFP-Spi-32R and RFP 0 min and 30 min after IPTG removal. (B) Spinach fluorescence as a function of time in the decay assay for RFP-Spi-32R, compared with the autofluorescence measured from E. coli expressing RFP. (C) RNA level in E. coli expressing RFP, RFP-Spi-8R and RFP-Spi-32R as a function of time in the decay assay, measured by qPCR.
As the Spinach array effectively enhanced the fluorescence signal compared to single Spinach tag and performed well in quantitatively reporting the mRNA abundance in live cells through imaging, we then tuned the mRNA transcription level to further characterize the imaging sensitivity of the Spinach array. We replaced the T7 promoter (PT7-RFP-Spi-32R) with a native lacZYA promoter (Plac) for RFP-Spi-32R transcription (Plac-RFP-Spi-32R). RNA synthesis by endogenous E. coli RNA polymerase instead of T7 polymerase reduced the mRNA expression level by approximately two orders of magnitude according to qPCR quantification (Fig. 4B). Using 16S ribosomal RNA (16S rRNA) (~20,000–70,000 copies per E. coli cell27,62) as a reference, we estimated the copy number of RFP-Spi-32R mRNA transcribed under the control of Plac and PT7 to be ~50–180 and ~3,000–11,000 per cell (Fig. 4B), respectively, which is consistent with the reported transcription levels for the two expression systems63,64. We first conducted epifluorescence microscopy on cells expressing Plac-RFP-Spi-32R with the same imaging condition applied for PT7-RFP-Spi-32R. Plac-RFP-Spi-32R did not show any fluorescence signal in Spinach channel beyond background autofluorescence level of uninduced Plac-RFP-Spi-32R (Supplementary Fig. S8), indicating that the epifluorescence microscope we used here is not sensitive enough to detect fluorescence signal from Spinach array with ~50–180 RNA copies per cell. In order to obtain higher fluorescence signal from the Spinach array, we used a 473 nm laser instead of the lamp light as the excitation source. A previous study showed that the DFHBI bound to the Spinach aptamer quickly dissociates (within ~100 ms) upon strong excitation, causing the loss of fluorescence43. To address the problem, we utilized pulsed excitation by laser to allow for Spinach to rebind a fluorogen after light-induced fluorogen dissociation and regain the fluorescence (Fig. 4A). Prior to imaging, we used continuous-wave (CW) illumination to decrease the cellular autofluorescence level (pre-photobeaching, or pre-PB) (Fig. 4A and Supplementary Fig. S9)65. Afterwards we used an automatically controlled mechanical shutter to generate a 0.2 Hz repetitive laser pulse with 50 ms pulse width43 (Supplementary Methods and Materials). With the help of pulsed illumination, we could observe clear distinction between uninduced Plac-RFP-Spi-32R (~4–15 mRNA copies per E. coli cell), where we hardly observed any fluorescence signal (Fig. 4D) and induced Plac-RFP-Spi-32R (~50–180 mRNA copies per E. coli cell), where we found bright spots in many cells residing at cell poles (Fig. 4C,D). We attribute the bright spots to transcription sites containing multiple mRNAs. The bright spots disappeared within a very short illumination time (<500 ms) and reoccurred upon illumination withdrawal (>5 s) followed by reinstatement (Fig. 4C), which is a typical optical characteristics of Spinach fluorescence43, suggesting that the fluorescence originated from the Spinach array. If we superposed multiple cell images from repeated cycles to achieve stronger fluorescence, we could clearly find preferential localization of the fluorescence at cell poles (Fig. 4D), consistent with the observed mRNA localization of induced Plac-RFP-Spi-32R verified by RNA FISH (Supplementary Fig. S10). Overall, with the help of the pulsed illumination method, we further enhanced the performance of the Spinach array and showed the potential to apply the system to image lower abundance cellular RNA.

(A) The pulsed illumination strategy to observe Spinach fluorescence, in which a 10 s continuous-wave (CW) illumination was applied to pre-photobleach (pre-PB) cellular autofluorescence, with a 10 s wait period after illumination withdrawal and then pulsed laser (power 0.2 mW, frequency 0.2 Hz, pulse duration 50 ms) was sent to illuminate the sample. (B) Expression level of RFP-Spi-32R mRNA under different promoters and induction conditions, measured by qPCR. (C) Representative fluorescence images of induced Plac-RFP-Spi-32R cells under CW or pulsed illumination. (D) Fluorescence images of induced and uninduced Plac-RFP-Spi-32R cells, shown in single frame (50 ms exposure time) or the superposition of 15 frames under pulsed illumination.
Discussion
The Spinach system is a recently developed RNA labeling and imaging method based on aptamer binding and fluorescence induction of the fluorogenic small molecule DFHBI34. It has many potential advantages over widely applied RNA labeling methods using fluorescent protein-fused RNA binding proteins, such as low fluorescence background, elimination of separate introduction of RNA binding proteins and evasion of perturbation on target RNAs by protein binding. However, there were few reports on single Spinach aptamer labeling for cellular mRNA imaging42, likely due to its low fluorescence brightness suggested by our experimental data (Fig. 2D). To address this problem, we employed a tandem Spinach array to tag a single mRNA molecule and demonstrated that the Spinach array containing 64 aptamer repeats can enhance the fluorescence by 17-fold in living cells compared to a single Spinach aptamer. The fluorescence enhancement by the Spinach array greatly improved mRNA imaging quality compared to using the single Spinach tag. In particular, we observed inhomogenous RNA distribution and distinct RNA loci in E. coli (Fig. 3D and Supplementary Fig. S4) using the Spinach array, whereas single Spinach tag suffered from low fluorescence signal and failed to report correct RNA localizations in cells. For cells with a lower mRNA level (~120–180 mRNAs per E. coli.), we further applied pulsed illumination strategy to effectively boost the fluorescence of the Spinach array such that we could observe mRNA localizations. Despite the 17-fold fluorescence enhancement achieved by constructing the aptamer array, we also noted that the average efficiency for an aptamer to correctly fold seems to decrease with the increase of the aptamer repeat number. One possible reason for this is the crosstalk and mispairing between adjacent or spatially close aptamers in a tandem array, which were reported and discussed for tandem arrays in previous RNA and protein folding studies51,52,53,66. In addition, the intrinsic instability of the Spinach aptamer may also play a role (~32% folded, 25 °C35). Future introduction of several recently reported aptamers with improved folding efficiency (Spinach 2, ~58%35; Broccoli, ~60%50) may enhance the fluorescence of the aptamer array. It is also possible that optimizing linker sequences may improve the performance although mispairing-based misfolding may not be avoided by changing linker sequences alone.
With a series of characterizations on the aptamer tandem array, we demonstrated that in E. coli, Spinach array-tagged mRNA had no significant alterations on transcription, translation or degradation. This may be attributed to the small size and high dissociation constant (KD) of the fluorogen. In contrast, the MS2/PP7 coat protein labeling method has been reported to impede RNA degradation in bacteria in previous studiess26,27, likely due to the stable association of many MS2/PP7 proteins to mRNA, which prevents the bacterial RNA degradation machinery from functioning. In addition, the Spinach array did not alter mRNA localization. When applied to eukaryotic cell imaging, in order to decrease the fluorescence background introduced by the MS2 coat protein-fluorescent protein fusion (MS2-FP) in cytosol, the MS2-FP proteins are usually fused with a nuclear localization sequence (NLS) to guide excess unbound proteins to the nucleus11,48. Although the strategy increases the signal-to-noise ratio, it might potentially perturb the endogenous RNA localization. In contrast, DFHBI remains non-fluorescent until binding to the Spinach aptamer, which circumvents extra modifications that decrease background fluorescence but could possibly affect target RNA localization.
In conclusion, we constructed a tandem Spinach aptamer array that could enhance fluorescence imaging quality of the Spinach/DFHBI system for live cell RNA imaging while introducing minimal perturbation to the target RNA. Nevertheless, Spinach arrays still show several limitations, including relatively low brightness and large size, making them ill-suited for studying mammalian cells, where autofluorescence is stronger and RNAs are more dispersedly distributed and undergo intensive motor-driven transport. Several recently developed fluorogen/aptamer systems, including RNA Mango, Spinach 2, Broccoli, etc, showed the potential to further enhance the performance. All of those newly discovered fluorogen/aptamer systems have similar fluorescing mechanisms as that of the Spinach aptamer while showing higher brightness, shorter length or more robust folding behavior. We envision that incorporating the newly reported aptamers into a tandem array could also enhance their fluorescence signals per RNA molecule. Furthermore, we expect the tandem arrays derived from those aptamers would be very likely to bring minimal perturbation to their target RNAs, due to their similarities to the Spinach aptamer.
Methods
The methods can be found in the Supplementary Information.
Additional Information
How to cite this article: Zhang, J. et al. Tandem Spinach Array for mRNA Imaging in Living Bacterial Cells. Sci. Rep. 5, 17295; doi: 10.1038/srep17295 (2015).
References
Alberts, B. et al. Molecular biology of the cell. fifth edn, (Garland Science, 2008).
Agrawal, N. et al. RNA interference: biology, mechanism and applications. Microbiol Mol Biol Rev 67, 657–685 (2003).
Martin, K. C. & Ephrussi, A. mRNA localization: gene expression in the spatial dimension. Cell 136, 719–730, doi: 10.1016/j.cell.2009.01.044 (2009).
Lecuyer, E. et al. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174–187, doi: 10.1016/j.cell.2007.08.003 (2007).
Rosenfeld, N., Young, J. W., Alon, U., Swain, P. S. & Elowitz, M. B. Gene regulation at the single-cell level. Science 307, 1962–1965, doi: 10.1126/science.1106914 (2005).
Tang, F., Lao, K. & Surani, M. A. Development and applications of single-cell transcriptome analysis. Nat Methods 8, S6–11, doi: 10.1038/nmeth.1557 (2011).
Li, G. W. & Xie, X. S. Central dogma at the single-molecule level in living cells. Nature 475, 308–315, doi: 10.1038/nature10315 (2011).
Raj, A., Van den Bogaard, P., Rifkin, S. A., Van Oudenaarden, A. & Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–879, doi: 10.1038/nmeth.1253 (2008).
Femino, A. M., Fay, F. S., Fogarty, K. & Singer, R. H. Visualization of single RNA transcripts in situ. Science 280, 585–590 (1998).
Hovelmann, F. et al. Brightness through local constraint—LNA-enhanced FIT hybridization probes for in vivo ribonucleotide particle tracking. Angew Chem Int Ed Engl 53, 11370–11375, doi: 10.1002/anie.201406022 (2014).
Tyagi, S. Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods 6, 331–338, doi: 10.1038/nmeth.1321 (2009).
Bao, G., Rhee, W. J. & Tsourkas, A. Fluorescent probes for live-cell RNA detection. Annual review of biomedical engineering 11, 25–47, doi: 10.1146/annurev-bioeng-061008-124920 (2009).
Wadia, J. S. & Dowdy, S. F. Protein transduction technology. Curr Opin Biotechnol 13, 52–56 (2002).
Giles, R. V. et al. Selecting optimal oligonucleotide composition for maximal antisense effect following streptolysin O-mediated delivery into human leukaemia cells. Nucleic Acids Res 26, 1567–1575 (1998).
Santangelo, P. J. et al. Single molecule-sensitive probes for imaging RNA in live cells. Nat Methods 6, 347–349, doi: 10.1038/nmeth.1316 (2009).
Belaya, K. & St Johnston, D. Using the mRNA-MS2/MS2CP-FP system to study mRNA transport during Drosophila oogenesis. Methods Mol Biol 714, 265–283, doi: 10.1007/978-1-61779-005-8_17 (2011).
Bertrand, E. et al. Localization of ASH1 mRNA particles in living yeast. Mol Cell 2, 437–445, doi: S1097-2765(00)80143-4 (1998).
Lionnet, T. et al. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat Methods 8, 165–170, doi: 10.1038/nmeth.1551 (2011).
Larson, D. R., Zenklusen, D., Wu, B., Chao, J. A. & Singer, R. H. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332, 475–478, doi: 10.1126/science.1202142 (2011).
Lim, F., Downey, T. P. & Peabody, D. S. Translational repression and specific RNA binding by the coat protein of the Pseudomonas phage PP7. J Biol Chem 276, 22507–22513, doi: 10.1074/jbc.M102411200 (2001).
Cilley, C. D. & Williamson, J. R. Analysis of bacteriophage N protein and peptide binding to boxB RNA using polyacrylamide gel coelectrophoresis (PACE). RNA 3, 57–67 (1997).
Valencia-Burton, M., McCullough, R. M., Cantor, C. R. & Broude, N. E. RNA visualization in live bacterial cells using fluorescent protein complementation. Nat Methods 4, 421–427, doi: 10.1038/nmeth1023 (2007).
Ozawa, T., Natori, Y., Sato, M. & Umezawa, Y. Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat Methods 4, 413–419, doi: 10.1038/nmeth1030 (2007).
Rath, A. K. & Rentmeister, A. Genetically encoded tools for RNA imaging in living cells. Curr Opin Biotechnol 31C, 42–49, doi: 10.1016/j.copbio.2014.07.012 (2014).
Wu, B., Chao, J. A. & Singer, R. H. Fluorescence fluctuation spectroscopy enables quantitative imaging of single mRNAs in living cells. Biophys J 102, 2936–2944, doi: 10.1016/j.bpj.2012.05.017 (2012).
Golding, I. & Cox, E. C. RNA dynamics in live Escherichia coli cells. Proc Natl Acad Sci USA 101, 11310–11315, doi: 10.1073/pnas.0404443101 (2004).
Golding, I., Paulsson, J., Zawilski, S. M. & Cox, E. C. Real-time kinetics of gene activity in individual bacteria. Cell 123, 1025–1036, doi: 10.1016/j.cell.2005.09.031 (2005).
Craggs, T. D. Green fluorescent protein: structure, folding and chromophore maturation. Chem Soc Rev 38, 2865–2875, doi: 10.1039/b903641p (2009).
Dean, K. M. & Palmer, A. E. Advances in fluorescence labeling strategies for dynamic cellular imaging. Nat Chem Biol 10, 512–523, doi: 10.1038/nchembio.1556 (2014).
Shin, I. et al. Live-cell imaging of Pol II promoter activity to monitor gene expression with RNA IMAGEtag reporters. Nucleic Acids Res 42, e90, doi: 10.1093/nar/gku297 (2014).
Babendure, J. R., Adams, S. R. & Tsien, R. Y. Aptamers switch on fluorescence of triphenylmethane dyes. J Am Chem Soc 125, 14716–14717, doi: 10.1021/ja037994o (2003).
Constantin, T. P. et al. Synthesis of new fluorogenic cyanine dyes and incorporation into RNA fluoromodules. Org Lett 10, 1561–1564, doi: 10.1021/ol702920e (2008).
Lee, J. et al. Combining SELEX screening and rational design to develop light-up fluorophore-RNA aptamer pairs for RNA tagging. ACS Chem Biol 5, 1065–1074, doi: 10.1021/cb1001894 (2010).
Paige, J. S., Wu, K. Y. & Jaffrey, S. R. RNA mimics of green fluorescent protein. Science 333, 642–646, doi: 10.1126/science.1207339 (2011).
Strack, R. L., Disney, M. D. & Jaffrey, S. R. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat Methods 10, 1219–1224, doi: 10.1038/nmeth.2701 (2013).
Dolgosheina, E. V. et al. RNA Mango Aptamer-Fluorophore: A Bright, High-Affinity Complex for RNA Labeling and Tracking. ACS Chem Biol, doi: 10.1021/cb500499x (2014).
Filonov, G. S., Moon, J. D., Svensen, N. & Jaffrey, S. R. Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J Am Chem Soc, doi: 10.1021/ja508478x (2014).
Hofer, K., Langejurgen, L. V. & Jaschke, A. Universal aptamer-based real-time monitoring of enzymatic RNA synthesis. J Am Chem Soc 135, 13692–13694, doi: 10.1021/ja407142f (2013).
Rogers, T. A., Andrews, G. E., Jaeger, L. & Grabow, W. W. Fluorescent Monitoring of RNA Assembly and Processing Using the Split-Spinach Aptamer. ACS Synth Biol. doi: 10.1021/sb5000725 (2014).
Paige, J. S., Nguyen-Duc, T., Song, W. & Jaffrey, S. R. Fluorescence imaging of cellular metabolites with RNA. Science 335, 1194, doi: 10.1126/science.1218298 (2012).
Song, W., Strack, R. L. & Jaffrey, S. R. Imaging bacterial protein expression using genetically encoded RNA sensors. Nat Methods 10, 873–875, doi: 10.1038/nmeth.2568 (2013).
Pothoulakis, G., Ceroni, F., Reeve, B. & Ellis, T. The spinach RNA aptamer as a characterization tool for synthetic biology. ACS Synth Biol 3, 182–187, doi: 10.1021/sb400089c (2014).
Han, K. Y., Leslie, B. J., Fei, J., Zhang, J. & Ha, T. Understanding the photophysics of the spinach-DFHBI RNA aptamer-fluorogen complex to improve live-cell RNA imaging. J Am Chem Soc 135, 19033–19038, doi: 10.1021/ja411060p (2013).
Kellenberger, C. A., Wilson, S. C., Sales-Lee, J. & Hammond, M. C. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J Am Chem Soc 135, 4906–4909, doi: 10.1021/ja311960g (2013).
Bhadra, S. & Ellington, A. D. A Spinach molecular beacon triggered by strand displacement. RNA 20, 1183–1194, doi: 10.1261/rna.045047.114 (2014).
Warner, K. D. et al. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat Struct Mol Biol 21, 658–663, doi: 10.1038/nsmb.2865 (2014).
Huang, H. et al. A G-quadruplex-containing RNA activates fluorescence in a GFP-like fluorophore. Nat Chem Biol 10, 686–691, doi: 10.1038/nchembio.1561 (2014).
You, M. & Jaffrey, S. R. Structure and Mechanism of RNA Mimics of Green Fluorescent Protein. Annu Rev Biophys 44, 187–206, doi: 10.1146/annurev-biophys-060414-033954 (2015).
Studier, F. W., Rosenberg, A. H., Dunn, J. J. & Dubendorff, J. W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 185, 60–89 (1990).
Filonov, G. S., Moon, J. D., Svensen, N. & Jaffrey, S. R. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J Am Chem Soc 136, 16299–16308, doi: 10.1021/ja508478x (2014).
Ha, T. Single-molecule approaches embrace molecular cohorts. Cell 154, 723–726, doi: 10.1016/j.cell.2013.07.012 (2013).
Borgia, M. B. et al. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature 474, 662–665, doi: 10.1038/nature10099 (2011).
Stigler, J., Ziegler, F., Gieseke, A., Gebhardt, J. C. & Rief, M. The complex folding network of single calmodulin molecules. Science 334, 512–516, doi: 10.1126/science.1207598 (2011).
Filonov, G. S., Kam, C. W., Song, W. & Jaffrey, S. R. In-gel imaging of RNA processing using broccoli reveals optimal aptamer expression strategies. Chem Biol 22, 649–660, doi: 10.1016/j.chembiol.2015.04.018 (2015).
Ponchon, L. & Dardel, F. Recombinant RNA technology: the tRNA scaffold. Nat Methods 4, 571–576, doi: nmeth1058 [pii] 10.1038/nmeth1058 (2007).
Nevo-Dinur, K., Nussbaum-Shochat, A., Ben-Yehuda, S. & Amster-Choder, O. Translation-independent localization of mRNA in E. coli. Science 331, 1081–1084, doi: 10.1126/science.1195691 (2011).
Nath, K. & Koch, A. L. Protein degradation in Escherichia coli II. Strain differences in the degradation of protein and nucleic acid resulting from starvation. J Biol Chem 246, 6956–6967 (1971).
Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S. & Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc Natl Acad Sci USA 99, 9697–9702, doi: 10.1073/pnas.112318199 (2002).
Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M. & Rosenow, C. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res 13, 216–223, doi: 10.1101/gr.912603 (2003).
Jach, G., Pesch, M., Richter, K., Frings, S. & Uhrig, J. F. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nat Methods 3, 597–600, doi: 10.1038/nmeth901 (2006).
Campbell, R. E. et al. A monomeric red fluorescent protein. Proc Natl Acad Sci USA 99, 7877–7882, doi: 10.1073/pnas.082243699 (2002).
Gourse, R. L., Gaal, T., Bartlett, M. S., Appleman, J. A. & Ross, W. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu Rev Microbiol 50, 645–677, doi: 10.1146/annurev.micro.50.1.645 (1996).
Miroux, B. & Walker, J. E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260, 289–298, doi: 10.1006/jmbi.1996.0399 (1996).
Terpe, K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72, 211–222, doi: 10.1007/s00253-006-0465-8 (2006).
Robin, F. B., McFadden, W. M., Yao, B. & Munro, E. M. Single-molecule analysis of cell surface dynamics in Caenorhabditis elegans embryos. Nat Methods 11, 677–682, doi: 10.1038/nmeth.2928 (2014).
Curtis, E. A. & Liu, D. R. A naturally occurring, noncanonical GTP aptamer made of simple tandem repeats. RNA Biol 11, 682–692 (2014).
Acknowledgements
We thank Prof. Samie R. Jaffrey for providing the pET28c-Spi-tRNA plasmid and Dr. Sung Chul Bae for providing the 473 nm laser. Funding was provided by NIH (GM065367, GM112659 and AI083025) and NSF (PHY0822613 and PHY1430124).
Author information
Authors and Affiliations
Contributions
J.Z., J.F. and T.H. designed the experiments. J.Z. performed the majority of the experiments and data analysis. J.F. performed some of the fluorometer measurement, epi-fluorescence imaging and quantitative PCR experiments. K.Y.H performed some of the fluorometer measurement and pulsed illumination imaging experiments. J.Z., J.F., B.J.L. and T.E.K. constructed the bacterial strains. B.J.L. synthesized the DFHBI. J.Z., J.F. and T.H. wrote the manuscript. T.H., J.Z., J.F., K.Y.H., B.J.L. and T.E.K. participated in the discussion.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, J., Fei, J., Leslie, B. et al. Tandem Spinach Array for mRNA Imaging in Living Bacterial Cells. Sci Rep 5, 17295 (2015). https://doi.org/10.1038/srep17295
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17295
This article is cited by
-
Engineered helicase replaces thermocycler in DNA amplification while retaining desired PCR characteristics
Nature Communications (2022)
-
Spinach-based RNA mimicking GFP in plant cells
Functional & Integrative Genomics (2022)
-
A protein-independent fluorescent RNA aptamer reporter system for plant genetic engineering
Nature Communications (2020)
-
Live cell imaging of single RNA molecules with fluorogenic Mango II arrays
Nature Communications (2020)
-
Nanomechanics and co-transcriptional folding of Spinach and Mango
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
