
Abstract
Persistent infection with oncogenic Human Papillomavirus (HPV) is necessary for cervical carcinogenesis. Although evidence suggests that the vaginal microbiome plays a functional role in the persistence or regression of HPV infections, this has yet to be described in women with cervical intra-epithelial neoplasia (CIN). We hypothesised that increasing microbiome diversity is associated with increasing CIN severity. llumina MiSeq sequencing of 16S rRNA gene amplicons was used to characterise the vaginal microbiota of women with low-grade squamous intra-epithelial lesions (LSIL; n = 52), high-grade (HSIL; n = 92), invasive cervical cancer (ICC; n = 5) and healthy controls (n = 20). Hierarchical clustering analysis revealed an increased prevalence of microbiomes characterised by high-diversity and low levels of Lactobacillus spp. (community state type-CST IV) with increasing disease severity, irrespective of HPV status (Normal = 2/20,10%; LSIL = 11/52,21%; HSIL = 25/92,27%; ICC = 2/5,40%). Increasing disease severity was associated with decreasing relative abundance of Lactobacillus spp. The vaginal microbiome in HSIL was characterised by higher levels of Sneathia sanguinegens (P < 0.01), Anaerococcus tetradius (P < 0.05) and Peptostreptococcus anaerobius (P < 0.05) and lower levels of Lactobacillus jensenii (P < 0.01) compared to LSIL. Our results suggest advancing CIN disease severity is associated with increasing vaginal microbiota diversity and may be involved in regulating viral persistence and disease progression.
Similar content being viewed by others

Introduction
Persistent infection with a high-risk oncogenic Human Papillomavirus (HPV) subtypes, most commonly 16 and 18, is a necessary, although not sufficient, condition for development of invasive cervical cancer (ICC) and its precancerous precursor; cervical intra-epithelial neoplasia (CIN)1. Although HPV infection is very common in sexually-active women2, the majority of infections are transient3. Only a small proportion of women infected with the virus goes on to develop clinically significant pre-invasive lesions and, if not treated, invasive malignant disease. Mechanisms of persistence of HPV infection are not well understood.
The vaginal microenvironment plays an important role in reproductive health. Commensal vaginal Lactobacillus spp. are thought to defend against pathogens and sexually transmitted infections4 through maintenance of a hostile pH5, production of species-specific metabolites, bacteriocins and through adherence to mucous and disruption of biofilms6,7,8,9. Next generation sequencing (NGS) based studies have facilitated detailed characterisation of the “healthy” vaginal microbiome and shown that 5 major community-state types (CSTs) exist; CST I, II, III and V are dominated by Lactobacillus crispatus, L. gasseri, L. iners and L. jensenii respectively, whereas CST IV has characteristically low numbers of Lactobacillus spp. and increased diversity of anaerobic bacteria10. Longitudinal studies of the vaginal microbiome using NGS indicates that bacterial community structure is dynamic and hormonally influenced with a propensity to become less stable during menstruation11 and conversely more stable and less diverse during normal pregnancy12,13. The stability and composition of the vaginal microbiome may play an important role in determining host innate immune response and susceptibility to infection. Bacterial vaginosis (BV), a condition characterised by Lactobacillus spp. depletion, overgrowth of anaerobic species and higher vaginal pH has been associated with increased transmission rates of sexually-transmitted infections14 and human immunodeficiency virus (HIV)15. Conversely, it has recently been reported that viral infection of the cervix during murine pregnancy increases susceptibility of ascending vaginal bacterial infection through sensitisation and priming of the host innate immune system16.
Relatively little is known about the mechanisms associated with clearance or persistence of HPV infection. Along with higher rates of HPV infection, BV has been associated with delayed clearance of the virus and with CIN, suggesting that a diverse, Lactobacillus-depleted microbiome may play a mechanistic role17,18,19,20. A recent study of 68 HPV-discordant monozygotic female Korean twins using NGS showed that HPV-positive twins had lower levels of Lactobacillus spp. and increased counts of Fusobacteria and Sneathia spp. compared to their HPV-negative twins21. Consistent with these findings, analysis of vaginal swabs collected longitudinally for 16 weeks from 32 sexually active women found that a Lactobacillus spp.-depleted, Atopobium spp. enriched (CST IV) community structure is associated with slowest regression of HPV whereas a Lactobacillus gasseri-dominated microbiome (CST II) is associated with the most rapid regression rates for HPV22.
While there is evidence of an association between vaginal microbiome structure and HPV infection, the potential relationship between the vaginal microbiota and CIN disease progression has yet to be investigated. In this study, we characterised the vaginal microbiome structure and diversity in women with pre-invasive cervical intraepithelial neoplasia, invasive cervical cancer and in healthy controls to assess how CSTs may correlate with disease presence and severity. We hypothesised that increasing microbiome diversity is associated with increasing CIN severity.
Results
We enrolled 169 women into the study who were classified into 4 groups; normal (n = 20), low-grade squamous intraepithelial lesion (LSIL) (n = 52), high-grade squamous intraepithelial lesion (HSIL) (n = 92) and ICC (n = 5). Table 1 shows the characteristics of each group. No difference in the mean age of the population was determined (mean 31, SD 5.08, range 23–45, P = 0.071). No category showed systematic bias with respect to the four disease-groups after adjustment for multiple hypothesis testing. There was equal distribution in the samples collected at the follicular or luteal phase of the cycle and in the rate of women that had intercourse within 48 hours from sample collection. Histology was available in all HSIL and cancer cases (100%) and for 69% (36/52) and 5% (1/20) of LSIL and normal cases, respectively.
The structure of the vaginal microbiome correlated to the disease severity
In total 8 409 192 reads were obtained from 169 samples with an average number of reads per sample of 49 759 and the mean and median read lengths of 513 and 520 bp respectively. To avoid sequencing bias, operational taxonomic units (OTUs) were randomly sub-sampled to the lowest read count of 183, which retained 97% of OTU counts (data not shown) and still provided coverage of >96% for all samples. Following removal of singletons and rare OTUs, a total of 49 taxa were identified in the vaginal microbiome of the study cohort.
Initial assessment of vaginal community structure was performed using principal component analysis (PCA) of species sequence data in the context of disease grade (normal, LSIL, HSIL and ICC) (Fig. 1). Three major clusters were identified which represented samples dominated by either L. crispatus, L. iners or samples depleted of Lactobacillus spp. with higher diversity. The results of the analysis at class level are presented in Supplementary Figure 1.

Bacterial species diversity in study cohort and controls.
Principle component analysis (PCA) of vaginal bacterial species data identified 3 major clusters corresponding to samples dominated by three community state types (CSTs): L. iners (CST III), L. crispatus (CST I) and high diversity samples (CST IV). KEY - Cancer: Black; High-grade squamous intra-epithelial lesions (HSIL): Red; Low-grade squamous intra-epithelial lesions (LSIL): Yellow; Normal: Green.
Hierarchical clustering analysis (HCA) of the sequence data using nearest neighbour linkage at species level (Fig. 2) identified 5 major clusters that exhibited bacterial community structure consistent with previously described vaginal microbiome community state types (CSTs); CST I: L. crispatus-dominated, CST II: L. gasseri-dominated, CST III: L. iners-dominated, CST IV: Lactobacillus-depleted and CST V: L. jensenii-dominated10 (Fig. 2). The results of the analysis at class level are presented in Supplementary Figure 2 & Supplementary Table 1.

Vaginal microbiome composition according to disease and HPV status.
Hierarchical clustering analysis (HCA) using centroid clustering showed the distribution of CSTs differs in healthy, normal control women (green), compared to those with disease (Disease Severity I). Frequency of CST IV was 2-fold greater in women with LSIL (yellow), 3-fold greater in HSIL (red) and 4-fold greater in women with ICC (black), compared to controls, with a reciprocal decrease in frequency of CST I with increasing disease severity. When HPV/ASCUS (light grey) and LSIL (dark grey) were examined as two separate groups, the stepwise increase in CST IV prevalence was maintained (Disease Severity II). HPV positivity (blue) was associated with increased rates of CST IV, compared to HPV negative women (light grey) who were most likely to have CST I (HPV status). HPV-16 (pink) was most frequently associated with CST IV (HPV genotype). Rates of CST IV were similar in women with normal or LSIL regardless of HPV status (negative = light grey, positive = dark grey) and were substantially higher in women with HSIL and positive for HPV (blue) (Disease severity I & HPV status). DISEASE SEVERITY I - Normal: Green; Low-grade squamous intra-epithelial lesions (LSIL): Yellow; High-grade squamous intra-epithelial lesions (HSIL): Red; Cancer: Black. DISEASE SEVERITY II - Normal: white; ASCUS: light grey; LSIL: dark grey; HSIL: blue; Cancer: pink. HPV STATUS - HPV negative: light grey; HPV positive: blue; Unknown: white. HPV GENOTYPE - HPV negative: light grey; HPV16: pink; HPV18: blue; Other HR-HPV: dark grey; Unknown: white. DISEASE SEVERITY I & HPV STATUS - LSIL/Normal HPV negative: light grey; LSIL/Normal HPV positive: dark grey; HSIL/HPV positive: blue; Cancer: pink; unknown/other: white. KEY - ASCUS: atypical squamous cells of undetermined significance; CST: community state type; HPV: Human Papillomavirus; HR-HPV: high-risk HPV; HSIL: High-grade squamous intra-epithelial lesion; ICC: invasive cervical cancer; LSIL: Low-grade squamous intra-epithelial lesion.
The rates and frequency of the different CSTs (I, II, III, IV & V) were compared between CIN disease severities and healthy controls (Table 2; Supplementary Table 3; Fig. 2). CST I was the most frequent CST in our study cohort (70/169, 41% of all patients), followed by CST III (47/169, 28%), CST IV (40/169, 24%), CST V (7/169, 4%) and CST II (5/169, 3%). Higher rates of CST IV (Lactobacillus-depleted, high diversity) were associated with increasing disease severity with CST IV observed twice as frequently in women with LSIL, three times as frequently in women with HSIL and four times as frequently in women with ICC when compared to disease free controls (normal = 2/20, 10%; LSIL = 11/52, 21%; HSIL = 25/92, 27%; cancer = 2/5, 40%). Conversely, frequency of CST I (Lactobacillus crispatus-dominant) was lower with increasing disease severity (normal = 10/20, 50%; LSIL = 22/52, 42%; HSIL = 37/92, 40%; cancer = 1/5, 20%). The number of ICC cases was small for any valid conclusion. Although the results of the analysis did not attain significance given the modest sample size, there appears to be a correlation of CST IV and increasing disease severity.
A similar distribution of CST IV was observed when women with HPV/Atypical squamous cells of undetermined significance (ASCUS) and LSIL changes were analysed as two separate groups (normal = 2/20, 10%; ASCUS = 5/26, 19%; LSIL = 6/26, 23%; HSIL = 25/92, 27%; cancer = 2/5, 40%) (Table 2; Fig. 2; Supplementary Table 3). These analyses are suggestive of association between CST IV and increasing disease severity and are consistent with the expected direction of effect, but do not attain significance due to modest sample-size.
Both species richness (Fig. 3A) and alpha-diversity (Fig. 3B,C) indices were higher in CST IV, compared to the other CSTs particularly CST I (P < 0.001) and CST III (P < 0.01) (Fig. 3). Consistent with increased rates of CST IV in high grade disease, vaginal microbiota richness and diversity were also found to be higher in women with high-grade disease, compared to low-grade disease and lowest in normal women but this was not statistically significant (Fig. 4) (Supplementary Table 4).

Analysis of richness (A) and diversity indices (B&C) with attributed CSTs for the patient cohort.
A significantly higher number of species observed in samples classified as CST IV (A) compared to CST I (P < 0.001) and CST III (P < 0.01). Diversity was also significantly higher in CST IV classified samples as assessed by the Inverse Simpson (B) and non-parametric Shannon (C) indices compared to CST I (P < 0.001) and CST III (P < 0.01). Kruskall-Wallis test (Dunn’s post hoc). KEY - CST: Community state type; Sobs: Species observed; ** = P < 0.01; *** = P < 0.001.

Vaginal microbiome richness and diversity indices associated with disease status (normal, LSIL, HSIL and cervical cancer patients).
The number of species observed increased with disease severity with lowest richness observed in healthy controls and highest in HSIL and ICC (A). Diversity, as assessed by Inverse Simpson (B) and non-parametric Shannon (C) indices followed the same pattern. KEY - HSIL: High-grade squamous intra-epithelial lesion; ICC: invasive cervical cancer; LSIL: Low-grade squamous intra-epithelial lesion; Sobs: Species observed; ** = P < 0.01; *** = P < 0.001.
The structure of the vaginal microbiome correlated to the HPV status and genotype
HPV status was available for 117 women in our cohort. CST IV was most frequently observed in high-risk HPV (HR-HPV) positive as compared to HR-HPV negative women (26/93, 28% versus 5/24, 21%). HPV negative women were most likely to have CST I (13/24, 54%) as opposed to HPV positive women (38/93, 41%). The rates for the other CSTs were comparable between HR-HPV positive and negative subjects (Table 2; Fig. 2).
Of 93 women who were HPV positive, genotyping was available for 62 subjects. The rate of CST IV was higher for women infected with HPV16 (9/31, 29%) when compared to HPV18 (1/5, 20%) or women with other high-risk oncogenic types (5/26, 19%) although did not reach significance, likely due to sample size (Table 2; Fig. 2).
The rate of CST IV was no different for Normal/LSIL HPV negative versus HPV positive individuals (3/20, 15% versus 7/34, 21%), but substantially higher for HSIL or worse (HSIL = 19/58, 33%; ICC = 2/5, 40%), suggesting that the presence of a high diversity Lactobacillus-depleted microbiome may be more strongly correlated to the presence of clinically significant pre- or invasive disease rather than the presence of the virus itself (Table 2; Supplementary Table 3; Fig. 2). Again, the results did not reach statistical significance.
Identification of vaginal microbiota composition markers of CIN disease severity
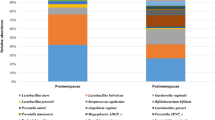
Linear discriminant analysis (LDA) effect size (LEfSe) modeling was used to identify differences in microbiota composition that may be related to increasing disease severity (Fig. 5). Due to sample size restrictions, we limited our comparison to LSIL versus HSIL patients. In the LSIL group, significant over-representation of Lactobacillus jensenii (P < 0.01) (Fig. 5A,B,F) and Lactobacillus coleohominis (P < 0.05) (Fig. 5B) were observed. In contrast, HSIL samples were found to have significantly higher levels of Peptostreptococcus anaerobius (P < 0.05) and Anaerococcus tetradius (P < 0.05) (Fig. 5A–D). HSIL samples were also found to have significant overrepresentation of Fusobacteria- primarily Sneathia sanguinegens (P < 0.01) (Fig. 5A,B,E; Supplementary Figure 3).

Identification of vaginal microbiota biomarkers of LSIL vs. HSIL by LEfSe analysis.
(A) Cladogram representing taxa with different abundance according to disease severity. Size of circle is proportionate to abundance of taxon. (B) Histogram of the LDA scores computed for features differentially abundant between LSIL and HSIL disease states. Relative abundance counts of Anaerococcus tetradius (C), Peptostreptococcus anaerobis (D) and Sneathia sanguinegens (E), which were found to be significantly over-represented in HSIL whereas Lactobacillus jensenii (F) was enriched in LSIL samples (Welch’s t-test). KEY - HSIL: High-grade squamous intra-epithelial lesion; LDA score: Linear discriminant analysis score; LSIL: Low-grade squamous intra-epithelial lesion.
Discussion
We detected a two-fold increase in the rate of a CST IV vaginal microbiome in those women with LSIL, a three-fold increase in women with HSIL and a four-fold increase in women with invasive cancer compared to controls. Increasing disease severity was also associated with decreasing relative abundance of Lactobacillus spp. A recent longitudinal study by Brotman et al. reported that women with a high diversity, Lactobacillus spp. depleted (CST IV) vaginal microbiome were most likely to become HPV-positive and to have persistent HPV infection22. Our findings suggest that vaginal microbial diversity is associated not only with HPV infection, but also with advancing CIN severity, but does not attain significance due to modest sample-size.
It is currently unclear if a CST IV microbiome is a causal factor in progression of CIN or a consequence of it. BV, a condition diagnosed using traditional culture techniques, in part by Lactobacillus spp. depletion and increased diversity of potentially pathogenic gram negative bacteria, is associated with significantly higher rates of HPV infection and CIN19,20. We used NGS techniques to further examine this association and identified Sneathia sanguinegens as a biomarker of HSIL, which has previously been shown to associate with HPV infection21. Two other BV-associated bacteria; Peptostreptococcus anaerobius and Anaerococcus tetradius were also found to be markers of HSIL in our cohort. This further suggests that specific anaerobic species may play a role in disease progression, rather than simply signifying presence of HPV infection.
Bacteria are increasingly appreciated as a key player in the initiation and progression of other malignancies including colorectal cancer23,24,25 where Fusobacteria has been identified as a potential pro-carcinogenic bacterial class25,26. Our findings show that this class and specifically Sneathia sanguinegens, is discriminatory of HSIL suggesting similar mechanisms, likely involving activation of inflammatory pathways, may be involved in the cervix. Approximately one third of premalignant lesions go on to develop invasive cervical disease, if untreated. It is possible that the women in our cohort with CST IV microbiomes are those at highest risk of progression to clinically significant invasive lesions, yet our findings only demonstrate association, not causality, between cervical pre-cancer, persistent HPV infection and the structure of the vaginal microbiota. Whilst women with BV display higher rates of HPV infection14,19 the virus has been shown to induce a pro-inflammatory environment to facilitate integration of viral DNA27,28,29,30,31. Thus HPV infection itself may adversely impact on the host’s immune defences and mucosal metabolism leading to aberration of vaginal microbiota, thus promoting viral persistence and disease progression.
Lactobacillus spp. are classically regarded as ‘protective’, yet our study supports other previous reports which suggest the clinical picture may be dictated by the specific species present and that the genus as a whole may not be regarded as protective in its entirety. Brotman and colleagues reported that an L. iners dominated vaginal microbiome (CST III) was associated with HPV-infection whereas vaginal microbiomes dominated by L. gasseri (CST II) exhibited the most rapid clearance of HPV infection22. While we did not observe over-representation of CST II or III in our CIN cohorts, the reduced prevalence of L. iners (CST III) concurrent with increased rates of CST IV in CIN, compared to controls may represents a shift from L. iners (CST III) towards a CST IV type microbiome with the acquisition of CIN. Unlike the majority of Lactobacillus spp., L. iners does not produce H2O2, which has been shown to have antibacterial and antiviral properties32,33,34. Consistent with the possibility that H2O2-producing Lactobacillus spp. are protective against CIN progression, we detected higher prevalence of L. jensenii and L. coleohominis, both H2O2-producing lactobacilli, in women with LSIL compared to HSIL, suggests this species may be particularly protective in preventing progression of the dysplastic and ultimately carcinogenic process. Furthermore Lactobacilli spp., have been shown to be cytotoxic when co-cultured with cervical cancer cells in vitro, but not normal cells, independent of lactic acid concentrations, highlighting interactions amongst cervical cells, the microbiota and the mucosal metabolic milieu35.
Environmental and hormonal factors are also known to modulate the vaginal microbiome. Smoking has been previously correlated to HPV persistence and CIN as well as Lactobacillus spp. depletion and dysbiosis36. Although women in our study with high-grade disease were more likely to be smokers, the differences did not reach statistical significance and no correlation between vaginal microbial composition and smoking status was identified. A sub-analysis of smokers showed that the prevalence of CST IV increased with disease severity indicating that a high-diversity microbiome is correlated to disease status rather than smoking as a potential confounder.
Future therapeutic strategies permitting the modulation of the vaginal microbiome with oral or topical regimes to a Lactobacillus spp.-dominant microbiome may be able to promote HPV clearance or even reverse the process of tumourigenesis, reducing the morbidity resulting from these conditions and their treatments37,38. Probiotics have been used in a similar manner to reduced recurrence of BV, through accurate, targeted modification of the bacterial community39.
Further research is required to understand the molecular mechanisms involved in the complex role that bacterial communities can play in the development of cancer. An understanding of the functional properties of the community state types is required in order to complement what we already know about their structure. Further longitudinal studies are needed to investigate the changes and stability of the microbiome during transition from acute HPV infection, to persistent infection through to development of CIN and cancer.
In summary, this is the first study to correlate the structure of the vaginal microbiome with presence of CIN in women of reproductive age. Our findings suggest that the presence and prevalence of specific vaginal microbiome CSTs may be involved in the pathogenesis of CIN and cervical cancer. We have also identified 5 bacterial species that could help to differentiate low- and high-grade disease and with further research these may improve our understanding of the role of the bacterial microenvironment in HPV persistence, development of CIN and progression to cancer. Although the development of HPV vaccines will be the main prevention strategy for this disease, its implementation in many settings can be challenging due to financial, cultural barriers and lack of infrastructure. Microbiome modulation with pre- and probiotics towards stable Lactobacillus-dominant vaginal community structure that promotes HPV clearance, could represent low-cost future therapeutic strategies.
Our findings may be of future clinical and therapeutic relevance and raise the clinical question as to whether women with a high diversity vaginal microbiome and cervical pathology should be subject to more intense colposcopic surveillance and/or treatment and whether the examination of the vaginal microbiome could be used as a triage tool for this population. Future longitudinal studies should aim to elucidate the causality between HPV infection, CIN, the immune microenvironment and the vaginal microbiome and increase our understanding of the role that vaginal bacteria in the tumour microenvironment.
Methods
Study population – Inclusion and Exclusion criteria
Ethical approval was obtained from the National Research Ethics Service Committee London – Fulham (Approval number 13/LO/0126). All experiments were performed in accordance with the approved guidelines. All patients gave informed consent. We included pre-menopausal non-pregnant women, 18–45 years of age who attended the colposcopy and gynaecology clinics at Imperial College NHS Healthcare Trust. Women were included irrespective of their ethnicity, parity, smoking habits, phase in their cycle and use of contraception. The type of contraception and the time of their cycle (follicular or luteal) were documented. Women who were HIV or hepatitis B/C positive, with autoimmune disorders, who received antibiotics or pessaries within 14 days of sampling, or had a previous history of cervical treatment were excluded. Detailed medical and gynaecological history was collected including time since last sexual intercourse and douching practices. Ethnicity was self-reported as Caucasian, Asian or Black. Histology was used to classify patients into groups. If histology was available from both punch biopsies and treatment cones, the most severe lesion was documented. If histology was not available as not clinically indicated (i.e. healthy controls, low-grade lesions under cytological surveillance), cytology was used for classification.
Sample collection and processing
A sterile, disposable speculum was inserted, without lubricant and a swab was taken from the posterior vaginal fornix using a BBLTM CultureSwabTM containing liquid Amies (Becton Dickinson, Oxford, UK) and stored immediately at −80 °C. Whole-Genomic bacterial DNA was extracted from the swabs using a QiAmp Mini DNA kit (Qiagen, Venlo, Netherlands) as previously described [12].
HPV genotyping
HPV DNA test and 16/18 genotyping was performed according to manufacturer’s guidelines using the Abbott RealTime High Risk (HR) HPV assay on Abbott M2000 platform; a clinically validated in vitro polymerase chain reaction (PCR) assay with identification of HPV-16, -18 and 12 other HR HPV subtypes (31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68)40.
Illumina MiSeq sequencing of 16S rRNA gene amplicons
The V1-V2 hypervariable regions of 16S rRNA genes were amplified by PCR using a forward and reverse fusion primer as previously described12. Sequencing was conducted at Research and Testing Laboratory (Lubbock, TX, USA).
16S rRNA gene sequence analysis
Sequence data were analysed in Mothur using the MiSeq SOP Pipeline41 Sequence reads were quality checked and normalised to the lowest number of reads. Singleton OTUs and OTUs < 10 reads in any sample were collated into OTU_singletons and OTU_rare phylotypes respectively, to maintain normalisation and to minimise artefacts. OTUs were defined using a cut off value of 97% and result data analysed using Vegan package within the R statistical package for assessment of microbial composition and diversity (R Development Core Team 2008). OTU taxonomies (from Phylum to Genus) were determined using the RDP MultiClassifier script to generate the RDP taxonomy42 while species level taxonomies of the OTUs were determined using the USEARCH algorithm combined with the cultured representatives from the RDP database43. Alpha and beta indices were calculated from these datasets with Mothur and R using the Vegan package.
Statistical analysis
Subjects were analysed in 4 different phenotype subgroups; normal, LSIL, HSIL and ICC. We included women with HPV changes or ASCUS in the LSIL category and further analysed them separately. Furthermore, we compared HR-HPV positive versus negative women, irrespective of the disease status and also assessed separately women positive for HPV16 versus HPV18 versus other high-risk (negative for HPV16 and HPV18) oncogenic subtypes. Finally, we used data from both the disease and HPV status and compared normal/LSIL HR-HPV negative women, normal/LSIL HR-HPV positive women, HSIL HR-HPV positive and ICC patients.
Analysis of statistical differences between the vaginal microbiome of patient groups was performed using the Statistical Analysis of Metagenomic Profiles (STAMP) package44. Data were subjected to multivariate analysis using PCA and HCA by nearest neighbour linkage with a clustering density threshold of 0.75.
To assess potential ascertainment bias of selected clinical characteristics with respect to four phenotype categories of interest, we performed fisher’s exact test for each of the following characteristics (age, ethnicity, parity, smoking, menstrual cycle, contraception and time since last intercourse). To analyse the importance of CSTs with respect to specific phenotype categories, we tested whether CSTs are significantly over or under-represented in any category. For this purpose we created a CST indicator variable, whereby CST = 1 for samples that could be assigned to the given CST and CST = 0 for all other samples. We performed a fisher’s exact test on the corresponding contingency table. Analyses were performed using R and false discovery rate adjustment (Benjamin & Hochberg) was applied to correct p-values. P-values and q-values < 0.05 were considered significant.
Additional Information
Accession code: Public access to sequence data and accompanying metadata can be obtained at the European Nucleotide Archive’s (ENA) Sequence Read Archive (SRA) (accession number PRJEB7756).
How to cite this article: Mitra, A. et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 5, 16865; doi: 10.1038/srep16865 (2015).
References
Munoz, N. Human papillomavirus and cancer: the epidemiological evidence. J. Clin. Virol. 19, 1–5 (2000).
Myers, E. R., McCrory, D. C., Nanda, K., Bastian, L. & Matchar, D. B. Mathematical model for the natural history of human papillomavirus infection and cervical carcinogenesis. Am. J. Epidemiol. 151, 1158–1171 (2000).
Richardson, H. et al. The natural history of type-specific human papillomavirus infections in female university students. Cancer Epidemiol. Biomarkers Prev. 12, 485–490 (2003).
Martin, D. H. The microbiota of the vagina and its influence on women’s health and disease. Am. J. Med. Sci. 343, 2–9, doi: 10.1097/MAJ.0b013e31823ea228 (2012).
Boskey, E. R., Cone, R. A., Whaley, K. J. & Moench, T. R. Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum. Reprod. 16, 1809–1813 (2001).
McMillan, A. et al. Disruption of urogenital biofilms by lactobacilli. Colloids Surf B Biointerfaces 86, 58–64, doi: 10.1016/j.colsurfb.2011.03.016 (2011).
Boris, S. & Barbes, C. Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect 2, 543–546 (2000).
Aroutcheva, A. et al. Defense factors of vaginal lactobacilli. Am. J. Obstet. Gynecol. 185, 375–379, doi: 10.1067/mob.2001.115867 (2001).
Ocana, V. S., Pesce De Ruiz Holgado, A. A. & Nader-Macias, M. E. Characterization of a bacteriocin-like substance produced by a vaginal Lactobacillus salivarius strain. Appl. Environ. Microbiol. 65, 5631–5635 (1999).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA 108 Suppl 1, 4680–4687, doi: 10.1073/pnas.1002611107 (2011).
Gajer, P. et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 4, 132ra152, doi: 10.1126/scitranslmed.3003605 (2012).
MacIntyre, D. A. et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 5, 8988, doi: 10.1038/srep08988 (2015).
Romero, R. et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2, 4, doi: 10.1186/2049-2618-2-4 (2014).
Brotman, R. M. et al. Bacterial vaginosis assessed by gram stain and diminished colonization resistance to incident gonococcal, chlamydial and trichomonal genital infection. J. Infect. Dis. 202, 1907–1915, doi: 10.1086/657320 (2010).
Atashili, J., Poole, C., Ndumbe, P. M., Adimora, A. A. & Smith, J. S. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 22, 1493–1501, doi: 10.1097/QAD.0b013e3283021a37 (2008).
Racicot, K. et al. Viral infection of the pregnant cervix predisposes to ascending bacterial infection. J. Immunol. 191, 934–941, doi: 10.4049/jimmunol.1300661 (2013).
Guo, Y., You, K., Qiao, J., Zhao, Y. & Geng, L. Bacterial vaginosis is conducive to the persistence of HPV infection. Int. J. STD AIDS 23, 581–584 (2012).
King, C. C. et al. Bacterial vaginosis and the natural history of human papillomavirus. Infect. Dis. Obstet. Gynecol. 2011, 319460, doi: 10.1155/2011/319460 (2011).
Gillet, E. et al. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: a meta-analysis. BMC Infect. Dis. 11, 10, doi: 10.1186/1471-2334-11-10 (2011).
Gillet, E. et al. Association between bacterial vaginosis and cervical intraepithelial neoplasia: systematic review and meta-analysis. PLoS One 7, e45201, doi: 10.1371/journal.pone.0045201 (2012).
Lee, J. E. et al. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One 8, e63514, doi: 10.1371/journal.pone.0063514 (2013).
Brotman, R. M. et al. Interplay Between the Temporal Dynamics of the Vaginal Microbiota and Human Papillomavirus Detection. J. Infect. Dis. doi: 10.1093/infdis/jiu330 (2014).
Marchesi, J. R. et al. Towards the human colorectal cancer microbiome. PLoS One 6, e20447, doi: 10.1371/journal.pone.0020447 (2011).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215, doi: 10.1016/j.chom.2013.07.007 (2013).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298, doi: 10.1101/gr.126573.111 (2012).
Viaud, S. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976, doi: 10.1126/science.1240537 (2013).
Hillier, S. L. & Lau, R. J. Vaginal microflora in postmenopausal women who have not received estrogen replacement therapy. Clin. Infect. Dis. 25 Suppl 2, S123–126 (1997).
Woodworth, C. D. HPV innate immunity. Front. Biosci. 7, d2058–2071 (2002).
Garcea, G., Dennison, A. R., Steward, W. P. & Berry, D. P. Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology 5, 514–529, doi: 10.1159/000087493 (2005).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867, doi: 10.1038/nature01322 (2002).
Scott, M., Stites, D. P. & Moscicki, A. B. Th1 cytokine patterns in cervical human papillomavirus infection. Clin. Diagn. Lab. Immunol. 6, 751–755 (1999).
Klebanoff, S. J. & Coombs, R. W. Viricidal effect of Lactobacillus acidophilus on human immunodeficiency virus type 1: possible role in heterosexual transmission. J. Exp. Med. 174, 289–292 (1991).
Klebanoff, S. J., Hillier, S. L., Eschenbach, D. A. & Waltersdorph, A. M. Control of the microbial flora of the vagina by H2O2-generating lactobacilli. J. Infect. Dis. 164, 94–100 (1991).
Clark, R. A. & Klebanoff, S. J. Role of the myeloperoxidase-H2O2-halide system in concanavalin A-induced tumor cell killing by human neutrophils. J. Immunol. 122, 2605–2610 (1979).
Motevaseli, E. et al. Normal and tumour cervical cells respond differently to vaginal lactobacilli, independent of pH and lactate. J. Med. Microbiol. 62, 1065–1072, doi: 10.1099/jmm.0.057521-0 (2013).
Brotman, R. M. et al. Association between cigarette smoking and the vaginal microbiota: a pilot study. BMC Infect. Dis. 14, 471, doi: 10.1186/1471-2334-14-471 (2014).
Kyrgiou, M. et al. Obstetric outcomes after conservative treatment for intraepithelial or early invasive cervical lesions: systematic review and meta-analysis. Lancet 367, 489–498, doi: 10.1016/S0140-6736(06)68181-6 (2006).
Kyrgiou, M. et al. Fertility and early pregnancy outcomes after treatment for cervical intraepithelial neoplasia: systematic review and meta-analysis. BMJ 349, g6192, doi: 10.1136/bmj.g6192 (2014).
Vujic, G., Jajac Knez, A., Despot Stefanovic, V. & Kuzmic Vrbanovic, V. Efficacy of orally applied probiotic capsules for bacterial vaginosis and other vaginal infections: a double-blind, randomized, placebo-controlled study. Eur. J. Obstet. Gynecol. Reprod. Biol. 168, 75–79, doi: 10.1016/j.ejogrb.2012.12.031 (2013).
Hesselink, A. T. et al. Clinical validation of the Abbott RealTime High Risk HPV assay according to the guidelines for human papillomavirus DNA test requirements for cervical screening. J. Clin. Microbiol. 51, 2409–2410, doi: 10.1128/JCM.00633-13 (2013).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120, doi: 10.1128/AEM.01043-13 (2013).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267, doi: 10.1128/AEM.00062-07 (2007).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, doi: 10.1093/bioinformatics/btq461 (2010).
Parks, D. H. & Beiko, R. G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721, doi: 10.1093/bioinformatics/btq041 (2010).
Acknowledgements
We thank all the participants of the study. Our work was supported by the British Society of Colposcopy Cervical Pathology (Jordan/Singer Award), the Imperial College Healthcare Charity, Genesis Research Trust and the Imperial Healthcare NHS Trust NIHR Biomedical Research Centre (P45272). DAM is supported by a Career Development Award from the Medical Research Council (MR/L009226/1).
Author information
Authors and Affiliations
Contributions
Designed project: A.M., D.A.M., E.P., P.R.B. and M.K. Collected samples: A.M., D.L. and M.K. Performed experiments: A.M., D.A.M, Y.L., R.B. and M.K. Analysed data: A.M., D.A.M., A.S., J.R.M., B.L. and M.K. Generated figures and tables: A.M., D.A.M., A.S., J.R.M., B.L. and M.K. Wrote manuscript: A.M., D.A.M. and M.K. All authors critically reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mitra, A., MacIntyre, D., Lee, Y. et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci Rep 5, 16865 (2015). https://doi.org/10.1038/srep16865
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16865
This article is cited by
-
Reconnoitering correlation between human papillomavirus infection-induced vaginal microecological abnormality and squamous intraepithelial lesion (SIL) progression
BMC Women's Health (2024)
-
The interplay between human papillomavirus and vaginal microbiota in cervical cancer development
Virology Journal (2023)
-
Characteristics of vaginal microbiota in various cervical intraepithelial neoplasia: a cross-sectional study
Journal of Translational Medicine (2023)
-
Changes in vaginal microbiome after focused ultrasound treatment of high-risk human papillomavirus infection-related low-grade cervical lesions
BMC Infectious Diseases (2023)
-
Optimal sampling and analysis methods for clinical diagnostics of vaginal microbiome
European Journal of Clinical Microbiology & Infectious Diseases (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
