
Abstract
The structural evolutionary behaviors of nitrogen in RbN3 have been studied up to 300 GPa using a particle swarm optimization structure searching method combined with density functional calculations. Three stable new phases with P-1, P6/mmm and C2/m structure at pressure of 30, 50 and 200 GPa are identified for the first time. The analysis of the crystal structures of three new predicated phases reveals that the transition of N3− ions goes from linear molecules to polymeric chains, benzene-like rings and then to polymeric layers induced by pressure. The electronic structures of three predicted phases reveal that the structural changes are accompanied and driven by the change of orbital hybridization of N atoms from sp to sp2 and finally to partial sp3. Most interestingly, the Rb atoms show obvious transition metal-like properties through the occupation of 4d orbitals in high-pressure phases. Moreover, the Rb atoms are characterized by strong hybridization between 4d orbitals of Rb and 2p orbitals of N in C2/m structure. Our studies complete the structural evolution of RbN3 under pressure and reveal for the first time that the Rb atoms in rubidium nitride possess transition element-like properties under pressure.
Similar content being viewed by others


Introduction
Metal azides have been the subjects of many studies including their structural stability, lattice dynamics, electronic structure and many other physical properties because of their linear azide anion1, as well as their significant industrial importance as gas generators and explosives2. Recently, taking metal azides as starting materials to synthesis polymeric nitrogen, a potential high-energy-density-material, has become a new topic due to the potential lower synthesis pressure compared with pure nitrogen gas. In nitrogen gas, nitrogen exits in N2 molecules and the connection between nitrogen atoms is triple bonds N≡N. In metal azides, nitrogen exits in  anions and the nitrogen atoms are connected through double bonds N = N. It is expected that the
anions and the nitrogen atoms are connected through double bonds N = N. It is expected that the  anions in metal azides may form a polymeric nitrogen network more readily than N2 molecules, since the N = N have a much lower bonding energy (418 KJ/mol) than the N≡N (954 KJ/mol). Under pressure,
anions in metal azides may form a polymeric nitrogen network more readily than N2 molecules, since the N = N have a much lower bonding energy (418 KJ/mol) than the N≡N (954 KJ/mol). Under pressure,  anion will undergo a series of structural transitions accompanied by the change of hybridization type of nitrogen atoms, as shown in Fig. 1. Under low pressure, usually fewer than 30 GPa,
anion will undergo a series of structural transitions accompanied by the change of hybridization type of nitrogen atoms, as shown in Fig. 1. Under low pressure, usually fewer than 30 GPa,  anions maintains their linear structures with sp hybridization, in which the crystal maybe undergo orientational phase transition of
anions maintains their linear structures with sp hybridization, in which the crystal maybe undergo orientational phase transition of  anions induced by pressure3,4,5,6,7. As pressure increase,
anions induced by pressure3,4,5,6,7. As pressure increase,  anions translates to a so called pseudo-benzene N6 ring with sp2 hybridization4,5,8,9. Continuing to increase pressure, nitrogen will form polymeric structure with partial sp3 hybridization. For CsN3, our previous work indicates that
anions translates to a so called pseudo-benzene N6 ring with sp2 hybridization4,5,8,9. Continuing to increase pressure, nitrogen will form polymeric structure with partial sp3 hybridization. For CsN3, our previous work indicates that  anions will translate to a chain like structure at 51 GPa instead of N6 ring10. In the process of nitrogen structural transition, alkali metal atoms in azides act as electronic donors to change the connection between nitrogen atoms and electronic properties of compounds. A lot of experimental and theoretical work has been done to study the high-pressure behaviors of nitrogen in LiN34,8,11,12,13,14,15, NaN33,5,16, KN37,9,14,17,18,19,20,21 and CsN310,22. Therefore, a study of the high-pressure behavior of RbN3 would provide more insights into the mechanism of pressure-induced structural evolution of
anions will translate to a chain like structure at 51 GPa instead of N6 ring10. In the process of nitrogen structural transition, alkali metal atoms in azides act as electronic donors to change the connection between nitrogen atoms and electronic properties of compounds. A lot of experimental and theoretical work has been done to study the high-pressure behaviors of nitrogen in LiN34,8,11,12,13,14,15, NaN33,5,16, KN37,9,14,17,18,19,20,21 and CsN310,22. Therefore, a study of the high-pressure behavior of RbN3 would provide more insights into the mechanism of pressure-induced structural evolution of  anions. It is helpful to investigate theoretically the pressure effect on rubidium azide and the role of rubidium atoms in the structural evolution process of
anions. It is helpful to investigate theoretically the pressure effect on rubidium azide and the role of rubidium atoms in the structural evolution process of  anions.
anions.

Under ambient conditions, α-RbN3 has the lowest energy with a body-centered tetragonal (bct) lattice of space group I4/mcm, in which rubidium (Rb), nitrogen 1 (N1) and nitrogen 2 (N2) atoms are located on the 4a,4d and 8 h Wyckoff positions, respectively (Fig. 2a),which is isostructural to the low-temperature phase of KN3 and CsN3 in all respects23. The nitrogen, linear and symmetric, occupy alternately [0,1,1] and [1  0] directed position in the crystal forming planes ((0 0 1) and (0 0 2) planes) separated by layers of Rb ions. Recently, for the high-pressure behaviors of RbN3, we present the in-situ X-ray diffraction studies of RbN3 up to 42.0 GPa at room temperature6. Two pressure-induced orientational phase transitions of α-RbN3 (I4/mcm) → γ-RbN3 (C2/m) → δ-RbN3 were identified at 6.5 and 16.0 GPa, respectively.
0] directed position in the crystal forming planes ((0 0 1) and (0 0 2) planes) separated by layers of Rb ions. Recently, for the high-pressure behaviors of RbN3, we present the in-situ X-ray diffraction studies of RbN3 up to 42.0 GPa at room temperature6. Two pressure-induced orientational phase transitions of α-RbN3 (I4/mcm) → γ-RbN3 (C2/m) → δ-RbN3 were identified at 6.5 and 16.0 GPa, respectively.

Crystal structures.
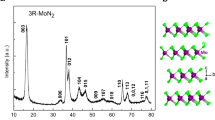
The ambient condition and the predicted high-pressure phase: (a) I4/mcm; (b) P-1; (c) P6/mmm; and (d) C2/m. The large and small spheres denote rubidium and nitrogen atoms, respectively. The red lines denote the connection between N6 rings in up section of (d). The nitrogen atoms in (a,b,d) have been labeled N1, N2 and N3 according to their Wyckoff positions.
In this work, we will focus on the structural evolution of anions under high pressure. A series of phase transitions in which  is converted to a chain like structure N6 ring and layered polymeric nitrogen have been discovered up to 300 G Pa by using a specifically developed particle swarm optimization (PSO) algorithm technique for crystal structure prediction24. To confirm the thermal dynamic stabilities of new phases, phonon dispersion spectroscopies have been calculated. The electronic properties calculations indicate the 4d orbitals of Rb have been partially occupied in high-pressure phases and a strong hybridization has been formed between 4d orbital of Rb and 2p orbital of N in layered structure of RbN3.
is converted to a chain like structure N6 ring and layered polymeric nitrogen have been discovered up to 300 G Pa by using a specifically developed particle swarm optimization (PSO) algorithm technique for crystal structure prediction24. To confirm the thermal dynamic stabilities of new phases, phonon dispersion spectroscopies have been calculated. The electronic properties calculations indicate the 4d orbitals of Rb have been partially occupied in high-pressure phases and a strong hybridization has been formed between 4d orbital of Rb and 2p orbital of N in layered structure of RbN3.
Computational Details
We have performed extensive structure searches to uncover the high-pressure structures of RbN3 based on a global minimization of free-energy surfaces merging ab initio total energy calculations via PSO technique, as implemented in the Crystal Structure Analysis by Particle Swarm Optimization (CALYPSO) code24,25. This method has successfully predicted the ground state structure for various systems including Nitrogen26, Caesium27 and superhard carbon nitride28. The underlying ab initio structural relaxations and electronic band structure calculations are performed in the framework of density functional theory within generalized gradient approximation Perdew-Burke-Ernzerhof (GGA-PBE)29, as implemented in the VASP code30. The projector augmented wave (PAW)31 pseudopotentials are adopted with the PAW potentials taken from the VASP library where 4p65s1 and 2s22p3 are treated as valence electrons for Rb and N atoms, respectively. The cutoff energy (800 eV) for the expansion of the wave function into plane waves and Monkhorst-Pack32 k-meshes (k-points density 0.03 Å−1) are chosen to ensure that all the enthalpy calculations are well converged to better than 1 meV/atom. The calculations of net charge are based on Bader analysis33,34. The phonon calculations are carried out by using a supercell approach as implemented in the PHONOPY code35.
Results and Discussion
The variable-cell high-pressure structure predictions have been performed within a pressure region from 0 to 300 GPa, with system containing from one to eight formula units per simulation cell as implemented in CALYPSO code. Our structural searches identified not only the ambient conditions phase I4/mcm shown in Fig. 2a, three new structures are also depicted in Fig. 2b,d. The lattice constants of predicted structure at ambient pressure a = 6.2871 Å and c = 7.5106 Å are in agreement with the results obtained in experiment (a = 6.3098 Å and c = 7.5188 Å23) which validates our computational method adopted here. The  ions are linear and symmetric and the bond length of N = N is 1.187 Å, which is same as that in potassium azides and well in agreement with the experimental results (1.176 Å23). The calculated atomic fractional coordinates are summarized in Table 1. The results indicate that
ions are linear and symmetric and the bond length of N = N is 1.187 Å, which is same as that in potassium azides and well in agreement with the experimental results (1.176 Å23). The calculated atomic fractional coordinates are summarized in Table 1. The results indicate that  anions undergo transition of
anions undergo transition of  → N chain → N6 ring → layered N. Different form converting directly into N6 ring structure in LiN34,8, NaN35 and KN37,9,
→ N chain → N6 ring → layered N. Different form converting directly into N6 ring structure in LiN34,8, NaN35 and KN37,9,  anions translate to chain-like structure, which appears in CsN310 at 51 GPa, before entering N6 ring structure. The layered nitrogen in C2/m phase is constructed by chair-like N6 rings, as shown in up section of Fig. 2d, completely different from the high-pressure structures in LiN34, NaN35 and KN37. In C2/m phase, nitrogen atoms have two nonequivalent sites 8j and 4i, respectively. The nitrogen atom located on 8j site is connected with three neighboring N through three N-N bonds and the N located on 4i site is connected with two neighboring N through two N-N bonds.
anions translate to chain-like structure, which appears in CsN310 at 51 GPa, before entering N6 ring structure. The layered nitrogen in C2/m phase is constructed by chair-like N6 rings, as shown in up section of Fig. 2d, completely different from the high-pressure structures in LiN34, NaN35 and KN37. In C2/m phase, nitrogen atoms have two nonequivalent sites 8j and 4i, respectively. The nitrogen atom located on 8j site is connected with three neighboring N through three N-N bonds and the N located on 4i site is connected with two neighboring N through two N-N bonds.
To investigate the energetic stabilities of RbN3 compound under high pressure, we calculate the formation enthalpy relative to the I4/mcm structure of RbN3 in a pressure range from 0 to 300 GPa, as shown in Fig. 3a. The most stable structure is a tetragonal phase with I4/mcm symmetry from ambient pressure which is then replaced by a lower-enthalpy P-1 structure at 30 GPa. Above 50 GPa, a hexagonal structure with P6/mmm symmetry is favored over other structures and remains the lowest-enthalpy phase up to 200 GPa. Continuously increasing pressure, the RbN3 will translate to a monoclinic structure with C2/m symmetry. Thorough structure searches using CALYPSO do not find any other structural change up to 300 GPa. The fact that the four structures are in entirely different crystal symmetry suggests that the transitions between them are first order which is indeed confirmed by the calculated P-V curves (Fig. 3b). The reductions of the volumes are found to be 10.1%, 1.9% and 3.7% for the transitions from I4/mcm to P-1, from P-1 to P6/mmm and from P6/mmm to C2/m, respectively.

(a) Enthalpy of formation of selected structures of RbN3 as a function of pressure (relative to the I4/mcm phase). (b) Phase diagram of RbN3 at pressure region from 0 to 300 GPa.
The dynamic stability of three predicted structures are examined by calculating the phonon spectra using the supercell method35. No imaginary phonon frequency is found in the whole Brillouin zone at the pressure 40 GPa, 100 GPa and 300 GPa, respectively, as shown in Fig. 4.

Phonon-dispersion curves of predicted structures.
(a) P-1, (b) P6/mmm and (c) C2/m at 40 GPa, 100 GPa and 300 GPa, respectively.
Under pressure, the structural evolution of the rubidium azide is accompanied by the change of the electronic properties. To explore that, we calculated the electronic structures and their dependence on pressure in several aspects, including the electron localized functions (ELF), band structures, electronic band structure, projected density of states (PDOS) and net charge of Rb atoms.
As shown in Fig. 5, the four phase transitions of RbN3 are accompanied by insulator-metal-metal-metal transitions. At ambient conditions, the I4/mcm structure is an insulator characterized by a large energy gap of 4.3 eV that is similar to the atmospheric pressure phases of other alkali metal azides7,10,12. However, the P-1, P6/mmm and C2/m structures exhibit clear metallic behaviors by evidence of cross of band structures and the finite electronic DOS at the Fermi level. As can be seen from the partial DOS, N-2p states contribute most to the valance band and the DOS near the Fermi level in P-1 and P6/mmm structures. The detailed analysis for metallicity of P-1 and P6/mmm can be found in previous works for CsN310 and KN37,9 due the similar electronic properties. For four considered structures, the strong covalent bonding between nitrogen atoms as well as the lone pairs electrons are revealed clearly by the ELF shown in Fig. 6. As pressure increase, the hybridization type between nitrogen atoms undergoes from sp (within I4/mcm) to sp2 (within P-1 and P6/mmm), then to partial sp3 (within C2/m).

Band structure and projected density of states.
(a) I4/mcm, (b) P-1, (c) P6/mmm and (d) C2/m at 0 GPa, 40 GPa, 100 GPa and 300 GPa, respectively. Dash line denotes Fermi energy level.

The electron localized functions.
(a) I4/mcm, (b) P-1, (c) P6/mmm and (d) C2/m at 0 GPa, 40 GPa, 100 GPa and 300 GPa, respectively. The valve of isosurface is 0.8.
More interestingly, the 4d orbital of rubidium is partially occupied in high-pressure phases as shown in Fig. 5b,d and there is an obvious orbital hybridization between the 4d of Rb and 2p of N located on 8j sites in C2/m phase. To further confirm this result, we recalculate the PDOS of the four phases using a much small Wigner-Seitz radius, in which we change the spherical radius from defaults value in pseudo-potential (2.418 and 0.741 Å for Rb and N) to smaller values based on the Bader analysis, as shown in supplementary Figure S1-S3. The recalculated results are similar to the previous PDOS for the four phases except the valve of densities. To study the impact of partial occupation of 4d orbital on electronic properties, we calculate the net charge of Rb atom based on Bader analysis, as shown in Fig. 7. At ambient conditions, the Rb atoms contribute almost one electron (0.85) to three N atoms forming  anion.
anion.

The net charge of Rb atoms based on Bader charge analysis for Rb in I4/mcm, P-1, P6/mmm and C2/m phases at 0 GPa, 40 GPa, 100 GPa and 300 GPa, respectively.
Under high pressure, the net charge decreases due to the partial occupation of 5d orbital, though the Rb atom still loses its 5s electron. Though the transition metal-like property of Rb elements has been reported by both experimental and theoretical works in alkali metal elements under high pressure36,37,38, there has been no research addressing that in chemical compounds. The difference that comes from the occupation of d orbitals is caused by the spd hybridization in alkali metal elements, for RbN3 that results from the hybridization between 4d of Rb and 2p of N which may enhance the stability of compound.
Conclusion
In summary, we studied the evolution of the structures of RN3 under high pressure by using an unbiased automatic structure search method based on first-principles total energy calculations and geometry optimization. We predicted three new high-pressure structures of RbN3 with P-1, P6/mmm and C2/m structure at pressure of 30, 50 and 200 GPa. This result extends the high-pressure structures RbN3. The analysis of the electronic structure reveals that the transition trend of  ions from linear molecules to polymer chains, then to benzene-like rings and finally to layered polymeric nitrogen is driven by the hybridization of N atoms in which the nitrogen hybridized types are sp, sp2, sp2 and partial sp3, respectively. For the first time, we reveal that the Rb atoms in RbN3 possess obvious transition metal-like properties under high pressure. In C2/m phase, a strong hybridization has been found between 4d orbitals of Rb and 2p orbitals of N.
ions from linear molecules to polymer chains, then to benzene-like rings and finally to layered polymeric nitrogen is driven by the hybridization of N atoms in which the nitrogen hybridized types are sp, sp2, sp2 and partial sp3, respectively. For the first time, we reveal that the Rb atoms in RbN3 possess obvious transition metal-like properties under high pressure. In C2/m phase, a strong hybridization has been found between 4d orbitals of Rb and 2p orbitals of N.
Additional Information
How to cite this article: Wang, X. et al. Layered polymeric nitrogen in RbN3 at high pressures. Sci. Rep. 5, 16677; doi: 10.1038/srep16677 (2015).
References
Fuith, A. The KSCN family: Structural properties and phase transitions of crystals with three-atomic linear anions. Phase Transitions A Multinatl. J. 62, 1–93 (1997).
Evans, B. L., Yoffe, A. D. & Gray, P. Physcis and chemistry of the inorganic azides. Chem. Rev. 59, 515–564 (1959).
Zhu, H. et al. Pressure-induced series of phase transitions in sodium azide. J. Appl. Phys. 113, 033511 (2013).
Wang, X. et al. Polymerization of nitrogen in lithium azide. J. Chem. Phys. 139, 1–6 (2013).
Zhang, M. et al. Structural and electronic properties of sodium azide at high pressure: A first principles study. Solid State Commun. 161, 13–18 (2013).
Li, D. et al. Pressure-induced phase transitions in rubidium azide: Studied by in-situ x-ray diffraction. Appl. Phys. Lett. 105, 071903 (2014).
Li, J. et al. Pressure-induced polymerization of nitrogen in potassium azides. EPL (Europhysics Lett.) 104, 16005 (2013).
Zhang, M., Yan, H., Wei, Q., Wang, H. & Wu, Z. Novel high-pressure phase with pseudo-benzene ‘N6’ molecule of LiN3 . EPL (Europhysics Lett.) 101, 26004 (2013).
Zhang, J. & Zeng, Z., Lin, H.-Q. & Li, Y.-L. Pressure-induced planar N6 rings in potassium azide. Sci. Rep. 4, 4358 (2014).
Wang, X., Li, J., Zhu, H., Chen, L. & Lin, H. Polymerization of nitrogen in cesium azide under modest pressure. J. Chem. Phys. 141, 044717 (2014).
Medvedev, S. a et al. Phase stability of lithium azide at pressures up to 60 GPa. J. Physics. Condens. Matter 21, 195404 (2009).
Babu, K. R., Lingam, C. B., Tewari, S. P. & Vaitheeswaran, G. High-pressure study of lithium azide from density-functional calculations. J. Phys. Chem. A 115, 4521–9 (2011).
Huang, X. et al. Large volume collapse during pressure-induced phase transition in lithium amide. J. Phys. Chem. C 116, 9744–9749 (2012).
Ramesh Babu, K., Vaitheeswaran, G. & Babu, K. R. Metal azides under pressure: An emerging class of high energy density materials. J. Chem. Sci. 124, 1391–1398 (2012).
Prasad, D. L. V. K., Ashcroft, N. W. & Hoffmann, R. Evolving Structural Diversity and Metallicity in Compressed Lithium Azide. J. Phys. Chem. C 117, 20838 (2013).
Eremets, M. I. et al. Polymerization of nitrogen in sodium azide. J. Chem. Phys. 120, 10618–23 (2004).
Ji, C. et al. High pressure X-ray diffraction study of potassium azide. J. Phys. Chem. Solids 72, 736–739 (2011).
Ramesh Babu, K. & Vaitheeswaran, G. Ab-initio study of structural and vibrational properties of KN3 under pressure. Chem. Phys. Lett. 533, 35–39 (2012).
Ji, C. et al. Pressure-induced phase transition in potassium azide up to 55 GPa. J. Appl. Phys. 111, 112613 (2012).
Hou, D. et al. Phase transition and structure of silver azide at high pressure. J. Appl. Phys. 110, 023524 (2011).
Hooper, J. & Zurek, E. High Pressure Potassium Polyhydrides: A Chemical Perspective. J. Phys. Chem. C 116, 13322–13328 (2012).
Hou, D. et al. Series of phase transitions in cesium azide under high pressure studied by in situ x-ray diffraction. Phys. Rev. B 84, 064127 (2011).
Müller, U. Crystal structure refinements of KN3, RbN3, CsN3 and TIN3 . Z. Anorg. Allg. Chem. 392, 159–166 (1972).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 82, 094116 (2010).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 183, 2063–2070 (2012).
Wang, X. et al. Cagelike Diamondoid Nitrogen at High Pressures. Phys. Rev. Lett. 109, 175502 (2012).
Miao, M.-S. Caesium in high oxidation states and as a p-block element. Nat. Chem. 5, 846–852 (2013).
Wang, X., Bao, K., Tian, F. & Meng, X. Cubic gauche-CN: A superhard metallic compound predicted via first-principles calculations. J. Chem. Phys. 133, 044512 (2010).
Perdew, J., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Monkhorst, H. J. & Pack, J. D. Special points for Brillonin-zone integrations. Phys. Rev. A 13, 5188–5192 (1976).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Tang, W., Sanville, E. & Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 21, 084204 (2009).
The program Phonopy is available at http://phonopy.sourceforge.net/; the force constant matrix is determined by VASP.
Parker, L. J., Atou, T. & Badding, J. V. Transition Element-Like Chemistry for Potassium Under Pressure. Science. 273, 95–97 (1996).
Ma, Y., Oganov, A. R. & Xie, Y. High-pressure structures of lithium, potassium and rubidium predicted by an ab initio evolutionary algorithm. Phys. Rev. B 78, 1–5 (2008).
Fabbris, G., Lim, J., Veiga, L. S. I., Haskel, D. & Schilling, J. S. Electronic and structural ground state of heavy alkali metals at high pressure. Phys. Rev. B 91, 1–9 (2015).
Acknowledgements
This work was supported by the National Science Foundation of China under Grants Nos. 11147007, 11304139, 11304111, 11274151, 11404278 and NSAF U1230202, Natural Science Foundation of Shandong Province No. ZR2014JL005 and Key Disciplines of Condensed matter Physics of Linyi University.
Author information
Authors and Affiliations
Contributions
X.W. and J.L. conceived the research. X.W. carried out the calculations. J.L., X.W., N.X., Z.H., H.Z. and L.C. analyzed the data. X.W. and J.L. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, X., Li, J., Xu, N. et al. Layered polymeric nitrogen in RbN3 at high pressures. Sci Rep 5, 16677 (2015). https://doi.org/10.1038/srep16677
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16677
This article is cited by
-
Formation mechanism of insensitive tellurium hexanitride with armchair-like cyclo-N6 anions
Communications Chemistry (2020)
-
Fe-N system at high pressure reveals a compound featuring polymeric nitrogen chains
Nature Communications (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
