
Abstract
The mesocrystal system fluoroapatite—gelatine grown by double-diffusion is characterized by hierarchical composite structure on a mesoscale. In the present work we apply solid state NMR to characterize its structure on the molecular level and provide a link between the structural organisation on the mesoscale and atomistic computer simulations. Thus, we find that the individual nanocrystals are composed of crystalline fluorapatite domains covered by a thin boundary apatite-like layer. The latter is in contact with an amorphous layer, which fills the interparticle space. The amorphous layer is comprised of the organic matrix impregnated by isolated phosphate groups, Ca3F motifs and water molecules. Our NMR data provide clear evidence for the existence of precursor complexes in the gelatine phase, which were not involved in the formation of apatite crystals, proving hence theoretical predictions on the structural pre-treatment of gelatine by ion impregnation. The interfacial interactions, which may be described as the glue holding the composite materials together, comprise hydrogen bond interactions with the apatite PO43− groups. The reported results are in a good agreement with molecular dynamics simulations, which address the mechanisms of a growth control by collagen fibers and with experimental observations of an amorphous cover layer in biominerals.
Similar content being viewed by others

Introduction
Mesocrystals or mesoscopically structured crystals are highly sophisticated materials, which are built up from individual nanocrystals as building blocks and which are aligned in a crystallographically ordered way1. Four principal possibilities are suggested for the remarkably ordered alignment and assembly of nanoparticles in mesocrystals2. These are an oriented organic matrix, the presence of electrostatic or magnetic fields, an epitaxial growth of nanoparticles with a mineral bridge between them and, finally, alignment by spatial constraints. Mesocrystals are much more common than assumed so far, but it is difficult to detect them, as they usually scatter X-rays like a single crystal and appear well-facetted3. Their unique controlled structure supports a unifying crystallization scenario, which combines both atomic/molecule-mediated (classical) and particle-mediated (non-classical) pathways4. Due to their unique combination of structural features related to high crystallinity and high porosity, as well as nanoparticles properties, mesocrystals provide solutions in many application areas. Recent progress on mesocrystals and their potential applications as heterogeneous photocatalysts, electrodes, optoelectronics, biomedical materials, hard templates, sensors and lightweight structural materials have been extensively reviewed in recent years5,6,7,8,9,10. Moreover, the possibility to tune the particle dimensions and the interactions between the physical and physicochemical properties of the individual nanoparticles by governing reaction conditions offers potential for developing novel materials with tailored functionalities. A natural process of biomineralization has also been found to take place via the particle-mediated pathway of mesocrystallization11,12. Since biominerals combine complex morphology and unique functional properties as the result of evolution-optimized processes, comprehensive insights into the biomineralization mechanisms open up promising approaches for bioinspired and biomimetic materials design11,13. In addition, adopting the bioinspired approach enables not only production of novel functional materials, but also better understanding of biomeralization processes through controlled synthesis and the in situ characterization methods. Recently, using a combination of in-situ methods the biomimetic nucleation of calcium phosphate, which is a major inorganic constituent of the hard tissues of animals, has been shown to occur through a unifying crystallization process14. Practical application of biomimetic strategies has been recently demonstrated for remineralization of human dentine15 and in vitro repair of a seashell16. It was suggested that organic macromolecules play important roles in the repair process and can control the mineralization reactions on several levels, including the formation of prenucleation clusters. In a recent review, Cölfen described the polymers that are useful for this purpose and the experimental conditions suitable for directing a crystallization reaction in the desired direction17.
However, a classical epitaxy paradigm, e.g., an epitaxial match between the structural organic matrix and the mineral, has been disproven by a HRTEM study of synthetic aragonite and of Haliotis laevigata gastropod nacre18,19. The existence of the amorphous layer around aragonite platelets in nacre is explained by the exclusion of impurities and expulsion of macromolecules throughout the crystallization, which prevents further crystallization. It has been suggested that this layer could also provide better adhesion and mechanical performance of the hybrid material. In our previous studies of synthetic hydroxy- and fluorapatite mesocrystals a disordered (amorphous) layer, which covers the apatite crystalline domains and is coordinated to water and the organic matrix, has been identified by solid-state NMR20,21. Thus, a disordered layer between aligned single crystalline nanoparticles embedded in an amorphous organic phase therefore seems to be an intriguing hypothesis for biominerals, which demonstrate mesocrystalline properties. Despite its relevance, little is known about the constituents and the molecular-level structure of this composite material. Therefore in the present work we continue our efforts on the structural elucidation of biomimetic fluoroapatite—gelatine nanocomposites.
The mesocrystal system fluoroapatite—gelatine grown by double-diffusion is a fair illustration of self-organized morphogenesis and a hierarchical structure on the mesoscale22,23,24,25,26,27,28. Growth of the nanocomposites starts with an elongated hexagonal prismatic seed (5–20 μm in length, Fig. 1a), followed by a self-similar branching of dumbbell states (Fig. 1b,c), which finally leads to the development of closed spheres. As proven by electron holography the fractal growth mechanism, which results in the dumbbell-like nanocomposite aggregates, is controlled by the intrinsic electrical dipole fields induced by an aligned polar biomolecule26,29. The inner architecture of the young seed, as indicated by high-resolution TEM, is built by a parallel stacking of elongated subunits oriented with their long [001] axes parallel to the seed. X-ray diffraction has revealed the presence of crystalline fluorapatite within the hexagonal prismatic seed, while a tilted mounting of the later crystal generations during the fractal morphogenesis has been observed30. On the mesoscopic scale, the so-called mosaic arrangement is suggested, where the periodic mineral domains of a nanocomposite subunit grow around a central protein triple-helix27. It has been shown that the domains do not perfectly match, giving rise to a healing layer at the outer borders to match the hexagonal pattern. As evidenced by electron microscopy, the gelatine molecules are surrounded by assemblies of fluorapatite nanoparticles arranged in a honeycomb-like network providing homogeneous intergrowth of the inorganic and organic components. Using solid state NMR we studied the interfacial mineral-organic structure in large spherical aggregates up to 100 μm in size, which represent the final growth state of the fractal-grown fluorapatite-gelatin composites20,21. As it is obvious that the fundamental principles of mineral growth, passivation and stabilization are already included in the early stages of composite growth, in the present work we focus on the study of the hexagonal prismatic seeds and dumbbells, thus the initial states of growth of fluorapatite/gelatin composites.

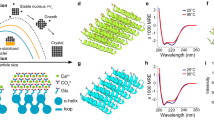
SEM images of fluorapatite-gelatine nanocomposite aggregates including (a) an elongated hexagonal-prismatic seed, (b) a dumbbell with initial splitting and (c) a “large” dumbbell with fractal branching.
Experimental section
Samples
Details of synthesis of fluorine-gelatine nanocomposites using the double diffusion technique have been published previously24,30,31. A composite aggregate extracted at the early growth stage was ground, washed 3 times for 20 min in distilled water at 40 °C, then centrifuged and finally dried at 40 °C in order to remove a fraction of gelatine, which is only physisorbed on the aggregates’ surface and not integrated in the composite.
X-Ray diffraction
X-ray powder data were collected in transmission mode using a Huber G670 Image Plate Camera, Cu Kα1 radiation (λ = 1.540598 Å) and germanium (111) monochromator. Lattice constants a and c of apatite were calculated by least-squares refinements using LaB6 (cubic, a = 4.15692 Å) as internal standard. The measurements were performed before and after heating to 250 °C to study the effect of release of hydroxyl groups and water from the crystal structure.
SEM
The morphology was studied by scanning electron microscopy (SEM). SEM investigations were performed by means of a Philips ESEM Quanta 200FEGI system operated in high-vacuum mode at an acceleration voltage of 25 kV (FEI, Eindhoven, NL). For investigation under high vacuum, the samples were coated by a thin gold layer (for 30 seconds) and secondary electron images were recorded.
NMR
For NMR experiments the samples were filled into zirconia rotors and closed tightly. 1H and 19F NMR experiments were carried out on a (11.7 T) Bruker Avance III 500 spectrometer operating at resonance frequencies of 500.1 MHz for 1H and 470 MHz for 19F using a 2.5 mm HFX-MAS probehead with a spinning frequency of 30 kHz. For 1H MAS NMR the 90°-pulse duration of 3 μs, a recycle delay of 5 s and high-power 19F TPPM (50 kHz) decoupling were applied. The 19F MAS NMR experiments were acquired with a 90°-pulse duration of 4.5 μs, a recycle delay of 40 s, a number of repetitions of 64 and TPPM (50 kHz) decoupling on protons. For the 19F{1H} cross polarization (CP) measurements contact times in the range of 100 μs to 1500 μs and a recycle delay of 5 s were used. The two-dimensional 19F-1H HETCOR experiment was performed at 30 kHz MAS and a contact time of 500 μs. A recycle delay of 5 s and 512 scans per t1 time increment were applied. A total of 32 t1 slices with a 50 μs time increment in the indirect dimension were acquired.
31P NMR spectra were acquired on a (7 T) Bruker Avance 300 spectrometer operating at resonance frequencies of 300.1 MHz for 1H and 121.5 MHz for 31P employing a BL4 HX 4 mm MAS probehead. The 31P MAS NMR experiments were performed at spinning frequencies of 10 and 14 kHz with either 1H or 19F high-power TPPM decoupling, a 90°-pulse duration of 4.5 μs, a recycle delay of 30 s and 16 repetitions. For 31P{1H} CP measurements a recycle delay of 5 s and a contact time varied from 100 μs to 4 ms were used. Two-dimensional 31P-1H HETCOR experiments were performed with a spinning speed of 10 kHz and contact times of 1.5 ms and 3 ms. A recycle delay of 5 s, 32 scans per t1 time increment and a 75.53 μs time increment in the indirect dimension were used. 31P{19F} CP MAS (10 kHz) spectra were measured at a contact time in the range of 0.1 ms to 4 ms, 16 repetitions and a recycle delay of 50 s.The 33P-19F HETCOR spectrum (10 kHz MAS) was acquired at a contact time of 1.5 ms, 64 scans per t1 time increment and a 200 μs time increment in the indirect dimension. All 1H chemical shifts were referenced to tetramethylsilane (TMS) at 0 ppm using poly(vinylidene fluoride) as an external reference (δ1H = 2.9 ppm); 19F chemical shifts were referenced relative to CFCl3 at 0 ppm using PTFE as an external reference (δ19F = −122 ppm); powdered ammonium dihydrogen phosphate was used to reference the 31P spectra at 0.72 ppm relative to 85% phosphoric acid. All spectra were fitted using Dmfit32.
Results
SEM
SEM images of the fluorapatite-gelatine composite demonstrate that the sample under study represents a combination of prismatic seeds (Fig. 1a) and dumbbell-like aggregates (Fig. 1b,c). According to the previously published results, this corresponds to earlier stages of morphogenesis, when an initially formed elongated hexagonal prismatic seed starts to grow and split, demonstrating fractal morphogenesis.
19F NMR
In the 19F MAS NMR spectrum of nanocomposite recorded by direct polarization (DP), five spectral components are identified (Fig. 2a), whose fit parameters are summarized in Table 1. The chemical shift values δiso19F for four lines are close to those found for the spherical aggregates reported in ref. 21. In the present study a minor high-field shoulder centred at ca. δiso19F = −86 ppm has been found in addition. Distribution of the component intensities essentially changes in the spectrum obtained by the cross-polarization (CP) showing different 1H-19F proximities of corresponding species (Fig. 2b). The peak at δ19F = −103 ppm is no longer visible in Fig. 2b, which indicates lack of spatial correlation to the protons and proves its assignment to pure crystalline fluorapatite21,33. Figure 2c shows the different CP build-up behaviour for the spectral components determined in the 19F{1H} CP MAS spectrum. In contrast to other signals, the 19F peak at −104 ppm continues growing up to tCP = 1 ms. Such behavior is characteristic of the crystalline apatite structure. Moreover, its chemical shift, which is very close to the pure crystalline fluorapatite, can indicate that the local apatite structure around this fluorine site is preserved34. Faster CP build-up of the remaining 19F signals, which reach a maximum at tCP = 0.1 ms (Fig. 2c), points to the close proximity to protons, perhaps, indicating covalent binding. In our previous work we attributed the 19F signal at ca. −109 ppm to an amorphous layer in close contact to the organic phase, while the signal around −96 ppm has been tentatively assigned to partial fluorination of gelatine during the formation of the nanocomposite21. Here we apply two-dimensional 19F-1H heteronuclear correlation (HETCOR) spectroscopy in order to analyse cross-correlation between the 1H and 19F signals and to get detailed information on the spatial association between 1H and 19F sites.

19F MAS NMR spectra detected using (a) direct polarization and (b) cross-polarization (CP) from 1H together with the deconvolution results.Spectra are normalized to a maximum intensity. (c) CP build-up as a function of the contact time tCP for four spectral components. Connecting lines are introduced for visualization.
The 19F-1H HETCOR spectrum (Fig. 3) demonstrates strong correlation to the peaks at δ1H = 8.2 ppm and δ1H = 5.8 ppm, which are attributed to certain molecular fragments of gelatin and water, respectively. The low-field shift (to higher ppm-values) relative to bulk water (δ1H = 4.8 ppm) is explained by strong hydrogen bonding of the water molecules to the organic molecule or orthophosphate groups. The crystalline fluorapatite peak at δ19F = −103 ppm also appears in the HETCOR spectrum, indicating the minor cross-signals at δ1H = 2.0 ppm and δ1H = 11.4 ppm. The former arises from isolated water molecules in the apatite channels and has been observed in the final growth stage21. The latter is assigned to HPO42− groups, which are frequently observed in the spectra of hydroxyapatite35 and other relevant systems, such as biological hard calcified tissues36,37 and biomimetic calcium phosphates21. The signals in the range of δ19F = −86 to −100 ppm cross-correlate to the 1H peaks related to water and the organic matrix.

19F-1H HETCOR spectrum with the corresponding sum-up projections.
31P NMR
The 31P MAS spectra are presented in Fig. 4. In contrast to the 31P MAS NMR spectrum of the spherical aggregates21, which represents a single fluorapatite 31P signal at δ31P = 2.8 ppm, in the early stage sample, in addition, a second component at δ31P = 2.1 ppm appears (Fig. 4a,b). Applying heteronuclear high-power decoupling enables linewidth narrowing due to suppression of the corresponding heteronuclear dipolar interactions. The effect of two different decoupling channels on the 31P spectrum (1H- and 19F-decoupled, Fig. 4a,b, respectively) is evidently indicating that the 31P site which gives a signal at 2.1 ppm is surrounded by a large proton bath. Indeed, this component is essentially amplified in the 31P spectrum recorded by cross-polarisation from protons (Fig. 4c) and nearly disappears when measured by cross-polarisation from 19F (Fig. 4d). The 31P{19F} CP spectrum (Fig. 4d) proves that the peak at 2.8 ppm arises from a pure fluorapatite structure21,23,24. In addition, a very low fraction of a non-apatite origin is visible in the 31P{1H} CP spectrum at 5.1 ppm. We can thus conclude that besides the dominating contribution from fluorapatite at δ31P = 2.8 ppm in the 31P spectra, there are small fractions of phosphorus-containing species spatially associated to protons.

31P MAS (10 kHz) NMR spectra detected using (a) direct polarization with high-power 1H decoupling, (b) direct polarization with high-power 19F decoupling, (c) cross-polarization from 1H and (d) cross-polarization from 19F together with the deconvolution results. Spectra are normalized to a maximum intensity.
Two-dimensional HETCOR experiments, where 31P chemical shifts are correlated to either 19F or 1H in the indirect dimension enable us to get deeper insight into the structural environment around phosphorous atoms. The 31P-19F HETCOR spectrum (Fig. 5a) demonstrates a single asymmetric cross-peak with a maximum intensity at δ31P = 2.8 ppm and δ19F = −103 ppm. This is a strong indication of the pure fluorapatite structure and support for the assignment given above. In the 19F sum projection a low-field shoulder at ca. −104 ppm is observed, which hints to the presence of another component not well separated in the 2D spectrum. Actually, its 19F line position and correlation to the peak at δ31P = 2.8 ppm points to its apatite origin. It is noteworthy that no other 31P-19F cross-peak is observed, demonstrating that the other peaks listed in Table 1 are of non-mineral origin at this growth stage.

(a) 31P-19F HETCOR and (b) 31P-1H HETCOR spectra with the corresponding sum-up projections.
In contrast, 31P-1H HETCOR shows a more complex spectrum (Fig. 5b). First, the cross-peaks associated with the apatite structure (δ31P = 2.8 ppm) are visible at δ1H = 5.8 ppm and δ1H = 11.4 ppm (dashed vertical line in Fig. 5b). They prove the presence of surface water and hydrogenated phosphate groups, respectively. Similar correlation signals have been observed in synthetic biomimetic nanocomposites20, as well as in biominerals such as joint mineralized cartilage38, animal bone39,40 and rat dentine41. Further, two strong cross-signals appear at δ31P/δ1H of 2.1 ppm/8.4 ppm and 5.0 ppm/8.4 ppm (Fig. 5b). According to the literature these 31P signals can be assigned to orthophosphate groups located at the mineral surface21,42,43 or to the side products of a double-diffusion reaction such as NaH2PO4H2O, K2HPO4· 3H2O and NaNH4HPO4· 4H2O, whose 31P signals are known to appear at 2.3, 2.1 and 5.1 ppm, respectively44. The signal at δ31P = 5.1 ppm has been observed in the one-dimensional 31P{1H} CP NMR spectrum (Fig. 4c) and attributed to non-apatite species. In either case, it is evident that these signals can arise from phosphate groups of non-apatite origin interacting with organic matrix. Note, that athough a low fraction of apatite-channel water has been found in 19F-1H HETCOR NMR (Fig. 3), the correlation signals at δ1H = 0 ppm and δ1H = 1.5 ppm, characteristic of apatite OH- groups and apatite channel water, respectively, are absent here.
Discussion
Fitting and quantification of the 19F MAS NMR spectrum obtained by direct polarization provides information about the relative fraction of each species resolved in the spectra. This allows us to get insight into the structure of the internal and interfacial regions of the early stage nanocomposite aggregates.
As suggested in our previous work for the final growth stage the mineral component is composed of crystalline apatite-like core surrounded by a disordered layer with first motifs of the apatite structure, which interacts with organic matrix21. In the earlier stage sample, the dominating contribution (72%) from all fluorine-containing species comes from crystalline fluorapatite (FAp) (Table 1, Fig. 2a). The 2D data (Fig. 3) show that FAp is associated weakly to HPO42- groups and isolated water molecules. The latter can be entrapped as structural defects and substitute fluorine ions in the apatite channels. The cross-peaks between FAp and gelatine/water molecules, which would prove the scenario of the periodic FAp domains’ growth around a protein triple-helix27,31, could be hidden beneath the large peaks due to their low fraction.
In the following, the origin of the 19F peak at −104 ppm is discussed. It represents 10% of all 1D 19F spectral intensity. Correlating this signal to the 31P dimension in the 2D spectrum (−104 ppm/2.8 ppm) proves its apatite character with the preserved local arrangement. However, long-range ordering is lacking as demonstrated by the line broadening as compared to crystalline fluorapatite (Fig. 2, Table 1). We anticipate that this species is located in a boundary layer, which “heals” the mismatch of the periodic domains to get a perfect hexagonal shape of the nanoparticles. Indeed, calculation of the intensity ratio for hexagonal geometry yields the thickness of such a layer of fractions of a nm, corresponding to the size of an orthophosphate molecule. Finding an identifiable species, which we will call a boundary layer in the following, is a clue to the hexagonal arrangement of primary apatite nanocrystals observed previously using HRTEM31. In general, the interfacial interactions, which may be described as the glue holding the composite materials together, may comprise nonspecific (e.g. van der Waals force driven alkane-surface interactions) and specific adsorption (hydrogen bonding, coulombic ion pairing) or covalent interfacial bridging45. Our 2D data (Fig. 3) show that the boundary layer interacts with gelatine and water. As the CP-build up data (green line in Fig. 2c) show that the corresponding fluorine-to-proton distances are too long for the formation of covalent bonds, we suggest that specific adsorption including hydrogen bonding to the apatite PO43− groups is the major interfacial interaction in the present organic-inorganic nanocomposite. The 31P-1H cross-peak at 2.8 ppm/5.8 ppm (Fig. 5b) can prove the presence of such hydrogen bonded complexes as PO43− ··· H(w) or PO43− ··· H(org). The appearance of the highly ordered hydrogen bond interactions between citrate and phosphate ions in spherulites of fluorapatite has been recently demonstrated using solid-state NMR46.
Furthermore, two phosphorous environments different from apatite are found in the 31P NMR spectra (Fig. 4) at δ31P = 2.1 and 5.1 ppm, whose fractions are relatively small compared to the whole phosphorous content. From the 2D HETCOR data it is evident that they are not related compositionally and spatially to FAp domains and the boundary layer (Fig. 5a), but related to the proton-containing organic phase (Fig. 5b). Moreover, the 1H cross-correlation signal at δ1H = 8 ppm proves association of the negatively charged phosphate groups to the positively charged amino groups. Thus, we suggest the existence of another spatially distinguishable phase, which contains organic matrix with incorporated fluorine ions, water molecules and isolated non-apatite phosphate groups. Indeed, our 2D HETCOR NMR measurements (Figs 3 and 5b) provide clear evidence for this claim. The absence of the corresponding peaks in the 31P-19F HETCOR experiment (Fig. 5a) is explained by a low concentration of the constituents, longer distances between phosphorous and fluorine atoms in this phase or their high mobility.
In the previous simulation study on this system47, which addressed the mechanisms of a growth control by collagen fibers, formation of Ca3F motifs has been postulated. According to ref. 47 such a motif is represented by a triangle formed by calcium ions, with a fluoride ion located in the center of the triangle. The Ca···F and Ca···Ca distances range from 2.0 to 2.5 Å and 3.6 to 4.7 Å, respectively and the angles of the triangles vary by up to 20° from the ideal value of 120°. These are incorporated into the triple helix during the embryonic stage of ion association. Such motifs represent the nucleation seeds for the formation of the oriented apatite crystal structure along the triple helix molecule. We assume here that the 19F signal observed at −108 ppm arise from Ca3F motifs, expected to occur during aggregation in/at the collagen triple helix. Indeed, its chemical shift is very close to that typical for Ca···F interactions, such as in crystalline CaF232, although no indication for the presence of CaF2 has been found in the corresponding X-ray powder diffraction data (Fig. S1, Supplementary information). It is worth noting that the presence of Ca3F and the absence of the corresponding CaF2 X-ray reflections have been also found for the spherical aggregates21. In contrast, the formation of calcium fluoride has been found in our previous studies of fluorapatite-gelatine composites, when gelatine concentration exceeded 15 wt.%.30, as well as more recently, when citric acid was used as a crystal modifier for the preparation of spherical hierarchical structures of FAp48. We emphasize that the present work is the experimental proof of the existence of Ca3F and PO43− precursor complexes in the gelatine phase, which were not involved in the formation of apatite crystals. Alternatively, the scenario that they were expelled from the single crystalline fluorapatite domain throughout crystallization as suggested in Refs.18,19 could also be feasible.
Our experimental observations are in a good agreement with previously published molecular dynamics simulations, which provide characteristic binding positions and preferable associations of the involved ions with a gelatine molecule47,49. It has been predicted that the phosphate ions are preferentially bound outside the polypeptide strands by forming hydrogen bonds with hydroxyproline side-groups and amino groups. This gives rise to bending of the polypeptide backbone and thus leads to the observed fractal morphogenesis. The same tendency has been predicted for HPO42− ion aggregation. Our NMR data provide clear evidence for the existence of such phosphate groups bound to organic molecules demonstrating the corresponding NMR signals. The 19F signals in the range of δ19F = −86 to −100 ppm are attributed to the amorphous organic-related phase, which contains fluorine ions hydrogen bonded to water or/and gelatine molecules.
Finally, when all species have been identified, it is worth considering the locations and interactions of water in the nanocomposite. In this work it has been found that (i) the isolated water molecules are incorporated as structural defects in the crystalline FAp domains; (ii) bound water is present on the mineral surface, and, finally, (iii) mobile water is included in the amorphous organic layer, where it strongly interacts with the gelatine molecules, non-apatite phosphate groups and Ca3F motives. Heating of the nanocomposite to 250 °C resulted in weight loss of 0.7 wt.% (Fig. S2, Supplementary information), demonstrating that most water molecules are integrated in the nanocomposite structure. The XRD pattern after heating (Fig. S1) proves no effect on the FAp crystalline structure.
Based on the experimental observations in the present work, a scheme of co-existing nano-structured mineral and a superficial organic layer in the fluorapatite-gelatine nanocomposite is presented in Fig. 6. The mineral part is represented by a mosaic arrangement of the periodic fluorapatite domains (grey circles) surrounded by the boundary layer (black circles), which matches the domains to the hexagonal shape and interacts with water and the organic layer impregnated with Ca3F and HPO42−/PO43− species.
Conclusions
A fluorapatite-gelatine nanocomposite at the early growth stage represented by hexagonal prismatic seeds and dumbbell-like aggregates with first sights of fractal splitting has been studied. We have applied solid-state NMR spectroscopy to investigate the structure in this nanocomposite on a molecular level and provide a link between electron microscopy data, electron holography and atomistic computer simulations. Based on our results we propose a model, which demonstrates the presence of a thin boundary layer around the crystallites and of pre-nucleation clusters in the amorphous organic-containing phase. The phosphate groups in the boundary layer are involved in the hydrogen bond interactions with the organic and water molecules in the amorphous layer. Our results are in a good agreement with the theoretical predictions on the structural pre-treatment of gelatine by ion impregnation and experimental observations of an amorphous cover layer in biominerals.
Additional Information
How to cite this article: Vyalikh, A. et al. An NMR Study of Biomimetic Fluorapatite – Gelatine Mesocrystals. Sci. Rep. 5, 15797; doi: 10.1038/srep15797 (2015).
References
Cölfen, H. & Antoniette, M. Mesocrystals and Nonclassical Crystallization (John Wiley & Sons Ltd. 2008).
Song, R. Q. & Cölfen, H. Mesocrystals—Ordered nanoparticle superstructures. Adv. Mater. 22, 1301–1330 (2010).
Revealed, I., Yuwono, V. M., Burrows, N. D., Soltis, J. A. & Lee Penn, R. Oriented aggregation: Formation and transformation of mesocrystal. J. Amer. Chem. Soc. 132, 2163–2165 (2010).
Cölfen, H. & Antonietti, M. Mesocrystals: Inorganic superstructures made by highly parallel crystallization and controlled alignment. Angew. Chem. Int. Ed. Engl. 44, 5576–5591 (2005).
Cho, K.-S., Talapin, D. V., Gaschler, W. & Murray, C. B. Designing PbSe nanowires and nanorings through oriented attachment of nanoparticles. J. Am. Chem. Soc. 127, 7140–7147 (2005).
Zhou, L. & O’Brien, P. Mesocrystals: A new class of solid materials. Small 4, 1566–1574 (2008).
Zhou, L. & O’Brien, P. Mesocrystals—Properties and applications. J. Phys. Chem. Lett. 3, 620–628 (2012).
Nie, Z., Petukhova, A. & Kumacheva, E. Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat. Nanotech. 5, 15–25 (2010).
Ma, M.-G. & Cölfen, H. Mesocrystals—Applications and potential (Review). Curr. Opin. Colloid Interface Sci. 19, 56–65 (2014).
Bu, F., Du, C. & Jiang, J. Synthesis, properties and applications of mesocrystals (Review). Progress in Chemistry 26, 75–86 (2014).
Meldrum, F. C. & Cölfen, H. Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 108, 4332–4432 (2008).
Politi, Y., Arad, T., Klein, E., Weiner, S. & Addadi, L. Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase. Science 306, 1161–1164 (2004).
Arakaki, A. et al. Biomineralization-inspired synthesis of functional organic/inorganic hybrid materials: Organic molecular control of self-organization of hybrids (Review). Org. Biomol. Chem. 13, 974–989 (2015).
Habraken, W. J. et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Comm. 4, 1507 (2013).
Tay, F. R. & Pashley, D. H. Guided tissue remineralisation of partially demineralised human dentine. Biomaterials 29, 1127–1137 (2008).
Kijima, M., Oaki, Y. & Imai, H. In vitro repair of a biomineral with a mesocrystal structure. Chem. Eur. J. 17, 2828–2832 (2011).
Cölfen, H. Polymer-mediated growth of crystals and mesocrystals. Methods in Enzymology 532, 277–304 (2013).
Nassif, N. et al. Synthesis of stable aragonite superstructures by a biomimetic crystallization pathway. Angew. Chem. Int. Ed. Engl. 44, 6004–6009 (2005).
Nassif, N. et al. Amorphous layer around aragonite platelets in nacre. Proc. Natl. Acad. Sci. 102, 12653–12655 (2005).
Vyalikh, A., Simon, P., Kollmann, T., Kniep, R. & Scheler, U. Local environment in biomimetic hydroxyapatite-gelatin nanocomposites as probed by NMR spectroscopy. J. Phys. Chem. C 115, 1513–1519 (2011).
Vyalikh, A. et al. Intergrowth and interfacial structure of biomimetic fluorapatite-gelatine nanocomposite: A solid-state NMR study. J. Phys. Chem. B 118, 724–730 (2014).
Busch, S. et al. Biomimetic morphogenesis of fluorapatite-gelatin composites. Fractal growth, the question of intrinsic electric fields, core/shell assemblies, hollow spheres and reorganization of denatured collagen. Eur. J. Inorg. Chem. 10, 1643–1653 (1999).
Göbel, C., Simon, P., Buder, J., Tlatlik, H. & Kniep, R. Phase formation and morphology of calcium phosphate-gelatine-composites grown by double diffusion technique: The influence of fluoride. J. Mater. Chem. 14, 2225–2230 (2004).
Kniep, R. & Simon, P. Fluorapatite-Gelatine-Nanocomposites. Self-Organized Morphogenesis, Real Structure and Relations to Natural Hard-Materials. In Topics in current chemistry (ed. Naka, K. ) 73–125 (Springer-Verlag, Berlin Heidelberg, 2007).
Simon, P., Schwarz, U. & Kniep, R. Hierarchical architecture and real structure in a biomimetic nanocomposite of fluorapatite with gelatine: A model system for steps in dentino- and osteogenesis? J. Mater. Chem. 15, 4992–4996 (2005).
Simon, P., Zahn, D., Lichte, H. & Kniep, R. Intrinsic electric dipole fields and the induction of hierarchical form developments in fluorapatite–gelatine nanocomposites. A general principle for morphogenesis of biominerals? Angew. Chem. Int. Ed. Engl. 45, 1911–1915 (2006).
Kniep, R. & Simon, P. "Hidden" hierarchy of microfibrils within 3D-periodic fluorapatite-gelatine nanocomposites: Development of complexity and form in a biomimetic system. Angew. Chem. Int. Ed. Engl. 47, 1405–1409 (2008).
Kniep, R. & Busch, S. Biomimetic growth and self-assembly of fluorapatite aggregates by diffusion into denatured collagen matrices. Angew. Chem. Int. Ed. Engl. 35, 2624–2626 (1996).
Simon, P., Rosseeva, E., Buder, J., Carrillo-Cabrera, W. & Kniep, R. Embryonic states of fluorapatite-gelatine nanocomposites and their intrinsic electric-field-driven morphogenesis. The missing link on the way from atomistic simulations to pattern formation on the mesoscale. Adv. Funct. Mater. 19, 3596–3603 (2009).
Busch, S., Schwarz, U. & Kniep, R. Chemical and structural investigations of biomimetically grown fluorapatite-gelatine composite aggregates. Adv. Funct. Mater. 13, 189–198 (2003).
Simon, P. et al. On the real-structure of biomimetically grown hexagonal prismatic seeds of fluorapatite-gelatine-composites. TEM investigations along [001]. J. Mater. Chem. 14, 2218–2224 (2004).
Massiot, D. et al. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 40, 70–76 (2002).
Braun, M. & Hartmann, P. 19F and 31P NMR spectroscopy of calcium apatites. J. Mater. Sci: Mater. in Medicine 6, 150–154 (1995).
Braun, M. & Jana, C. 19F NMR spectroscopy of fluorinated apatites. Chem. Phys. Lett. 245, 19–22 (1995).
Yesinowski, J. P. & Eckert, H. Hydrogen environments in calcium phosphates—H-1 MAS NMR at high spinning speeds. J. Amer. Chem. Soc. 109, 6274–6282 (1987).
Vyalikh, A., Mai, R. & Scheler, U. OH deficiency in dental enamel, crown and root dentine as studied by 1H CRAMPS. Bio-Med. Mater. Eng. 23, 507–512 (2013).
Kolmas, J. & Kolodziejski, W. Concentration of hydroxyl groups in dental apatites. a solid-state H-1 MAS NMR study using inverse P-31 ->H-1 cross-polarization. Chem. Commun. 42, 4390–4392 (2007).
Duer, M. J., Friscic, T., Murray, R. C., Reid, D. G. & Wise, E. R. The mineral phase of calcified cartilage. Its molecular structure and interface with the organic matrix. Biophysical J. 96, 3372–3378 (2009).
Wilson, E. E. et al. Three structural roles for water in bone observed by solid-state NMR. Biophysical J. 90, 3722–3731 (2006).
Santos, R. A., Wind, R. A. & Bronnimann, C. E. 1H CRAMPS and 1H-31P HetCor experiments on bone, bone mineral and model calcium phosphate phases. J. Magn. Reson. B 105, 183–187 (1994).
Tseng, Y.-H. et al. Characterization of the phosphate units in rat dentin by solid-state NMR spectroscopy. Chem. Mater. 19, 6088–6094 (2007).
Jarlbring, M., Sandstrom, D. E., Antzutkin, O. N. & Forsling, W. Characterization of active phosphorus surface sites at synthetic carbonate-free fluorapatite using single-pulse H-1, P-31 and P-31 CP MAS NMR. Langmuir 22, 4787–4792 (2006).
Wu, Y., Glimcher, M. J., Rey, C. & Ackerman, J. L. A unique protonated phosphate group in bone mineral not present in synthetic calcium phosphates. Identification by phosphorus-31 solid state NMR spectroscopy. J. Mol. Biol. 244, 423–435 (1994).
Turner, G., Smith, K. A., Kirkpatrick, R. & Oldfield, E. Structure and cation effect on Phosphorus-31 NMR chemical shift and chemical-shift anisotropies of orthophosphates. J. Magn. Reson. 70, 408–415 (1986).
Texter, J. Interfaces in advanced materials. Curr. Opin. Colloid. In. 19, 43–48 (2014).
Wu, Y.-J. et al. Hydrogen bond formation between citrate and phosphate ions in spherulites of fluorapatite. Langmuir 29, 11681–11686 (2013).
Kawska, A., Hochrein, O., Brickmann, J., Kniep, R. & Zahn, D. The nucleation mechanism of fluorapatite-collagen composites. Ion association and motif control by collagen proteins. Angew. Chem. Int. Ed. Engl. 47, 4982–4985 (2008).
Wu, Y.-J., Tseng, Y.-H. & Chan, J. Morphology control of fluorapatite crystallites by citrate ions. Crystal Growth & Design 10, 4240–4242 (2010).
Tlatlik, H., Simon, P., Kawska, A., Zahn, D. & Kniep, R. Biomimetic fluorapatite-gelatine nanocomposites: Pre-structuring of gelatine matrices by ion impregnation and its effect on form development. Angew. Chem. Int. Ed. Engl. 45, 1905–1910 (2006).
Acknowledgements
We cordially thank Dr. Robert Carley for editing the manuscript. E.S. thanks the Deutsche Forschungsgemeinschaft (RO 4511/1-1) and the Zukunftskolleg at the University of Konstanz for financial support. A.V. acknowledges Prof. Hermann Ehrlich and Prof. Dirk C. Meyer for fruitful suggestions and discussions.
Author information
Authors and Affiliations
Contributions
P.S. and E.R. prepared the samples and characterized them. J.B. prepared figures S1 and S2. A.V. and U.S. performed NMR experiments. A.V., P.S., J.B. and R.K. discussed the results. R.K. contributed to Figure 6. The manuscript was written by A.V. and P.S. All authors reviewed the manuscript.

Sketch of the co-existing nano-structured mineral and organic components in the fluorapatite nanocomposite.
The mineral part is represented by a mosaic arrangement of the periodic crystalline FAp domains (grey circles) surrounded by a boundary layer (black circles), which interacts with the surface water and the organic layer. The organic component incorporates Ca3F motifs, phosphate groups, fluorine ions and water molecules.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vyalikh, A., Simon, P., Rosseeva, E. et al. An NMR Study of Biomimetic Fluorapatite – Gelatine Mesocrystals. Sci Rep 5, 15797 (2015). https://doi.org/10.1038/srep15797
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15797
This article is cited by
-
Bone mineral: new insights into its chemical composition
Scientific Reports (2019)
-
Green self-assembly of zein-conjugated ZnO/Cd(OH)Cl hierarchical nanocomposites with high cytotoxicity and immune organs targeting
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
