
Abstract
Mesenchymal stem cells (MSCs) have regenerative properties, but recently they were also found to have immunomodulatory capacities. We therefore investigated whether MSCs could reduce atherosclerosis, which is determined by dyslipidaemia and chronic inflammation. We adoptively transferred MSCs into low-density lipoprotein-receptor knockout mice and put these on a Western-type diet to induce atherosclerosis. Initially after treatment, we found higher levels of circulating regulatory T cells. In the long-term, overall numbers of effector T cells were reduced by MSC treatment. Moreover, MSC-treated mice displayed a significant 33% reduction in circulating monocytes and a 77% reduction of serum CCL2 levels. Most strikingly, we found a previously unappreciated effect on lipid metabolism. Serum cholesterol was reduced by 33%, due to reduced very low-density lipoprotein levels, likely a result of reduced de novo hepatic lipogenesis as determined by a reduced expression of Stearoyl-CoA desaturase-1 and lipoprotein lipase. MSCs significantly affected lesion development, which was reduced by 33% in the aortic root. These lesions contained 56% less macrophages and showed a 61% reduction in T cell numbers. We show here for the first time that MSC treatment affects not only inflammatory responses but also significantly reduces dyslipidaemia in mice. This makes MSCs a potent candidate for atherosclerosis therapies.
Similar content being viewed by others


Introduction
Mesenchymal stem cells (MSCs), also called bone marrow stromal cells or mesenchymal stromal cells, are multipotent cells that can give rise to cells of the mesodermal lineage, including adipocytes, osteocytes and myocytes1. They were first identified in the bone marrow, but can be isolated from other tissues such as umbilical cord, placenta and adipose tissue2. After isolation, MSCs can be easily expanded without losing their multipotency which renders them an interesting tool for therapeutic strategies1. MSCs were initially investigated for their ability to repair injured heart tissue after myocardial infarction3,4,5. They can migrate to sites of tissue damage and inflammation, where they can extravasate, engraft the tissues and reduce scar formation4,6,7,8,9.
In recent years the immunomodulatory capacity of MSCs has been increasingly appreciated. Several studies have investigated the capacity of MSCs to modulate both innate and adaptive immune responses2,10,11,12,13,14,15. For instance, MSCs have been shown to reduce monocyte responses after myocardial infarction11 and to skew macrophages to an anti-inflammatory IL-10-producing phenotype11,12,13,14. MSCs also inhibit the differentiation and maturation of dendritic cells (DCs)15, by reducing the expression of co-stimulatory molecules and pro-inflammatory cytokines (TNF-α and IL-12), while increasing the production of anti-inflammatory cytokines (TGF-β and IL-10)16,17, which indirectly suppresses T cell proliferation17. However, MSCs can also directly inhibit T cell proliferation16,17,18, by inducing cell cycle arrest in all subsets, resulting in a quiescent state and decreased proliferation19.
Inflammatory processes play a crucial role in all stages of atherosclerosis. Early in the disease process, entrapped oxidized low-density lipoprotein (oxLDL) in the vessel wall leads to the activation of arterial endothelial cells and an ensuing recruitment of monocytes and T cells20. Upon recruitment, monocytes can differentiate to macrophages21. Macrophages can primarily promote T cell responses to local antigens, while DCs can activate naïve T cells in response to lesion-derived antigens in draining lymph nodes22. DCs are also present within lesions or can arise from blood-derived precursors. Both macrophages and DCs express scavenger receptors enabling the uptake of oxLDL and foam cell formation and toll-like receptors, which mediate activation of the antigen-presenting cells and production of pro-inflammatory cytokines. CD4+ T cells are crucially involved in the pathogenesis of atherosclerosis and their depletion results in a 70% reduction of lesion size23. The predominant subset in both human and murine atherosclerotic lesions is the Th1 subset24,25, which produces a plethora of pro-inflammatory cytokines such as IFN-γ. IFN-γ promotes vascular inflammation by enhancing maturation and activation of antigen-presenting cells, increasing macrophage lipid uptake, reducing collagen production by smooth muscle cells and enhancing expression of endothelial adhesion molecules, which subsequently stimulates leukocyte recruitment to the lesions26. The continuous recruitment of further leukocytes to atherosclerotic lesions results in a vicious self-maintaining progressive inflammatory cycle. Nonetheless, it has been shown that by altering the phenotype of macrophages and DCs, they can become atheroprotective22,27,28. Moreover, regulatory T cells (Tregs) have been clearly established as anti-atherogenic29. Tregs produce high amounts of the anti-inflammatory cytokine IL-10 and inhibit ongoing inflammation.
Due to the key role of inflammatory processes in the initiation and progression of atherosclerosis, adoptive transfer of MSCs, which have the capacity to modulate and reduce inflammation, may be a therapeutic approach to treat atherosclerosis. Preclinical studies have already shown that adoptively transferred MSCs are capable of modulating immune responses and they can prevent allograft rejection18,30 and alleviate autoimmune diseases31,32,33. Moreover, in a phase II clinical trial, it was found that MSCs can reduce graft-versus-host disease34. We now show to our knowledge for the first time that MSCs in addition to their immunomodulatory capacity can significantly reduce dyslipidemia, thus establishing the multifactorial therapeutic potential of MSCs to inhibit atherogenesis.
Results
MSCs were generated from the bone marrow of male C57BL/6 mice and their phenotype was confirmed by flow cytometry analysis. All MSCs expressed Sca-1, CD29, CD44, CD105 and CD106, while CD45, CD31 and TER119 were not expressed (Supplementary Fig. 1). To confirm their immunomodulatory capacity, MSCs were co-cultured with DCs for three hours at different ratios and subsequently stimulated with LPS. Twenty-four hours after LPS stimulation, we did not observe effects of MSCs on the expression of the co-stimulatory molecules CD40, OX40L and CD30L by DCs. The percentage of DCs positive for the co-stimulatory molecule CD86 and the negative co-stimulatory molecule PD-L2 increased by 12% and 22%, respectively. The mean expression of CD86 per cell was not significantly affected, whereas the mean expression of PD-L2 was decreased by 18% at higher MSC to DC ratios. The mean expression of the co-stimulatory molecule CD80 on DCs was significantly increased by 34% (Table 1; P < 0.001) upon co-culture with MSCs. CD80/CD86 function to induce T cell activation but can also promote Treg development, while PD-L2 functions to inhibit T cell activation, indicating that DCs might adopt a more tolerogenic phenotype after exposure to MSCs. In line with this, MSCs significantly affected the cytokine production of DCs in response to LPS. Pro-inflammatory TNF-α release was reduced by 57% (P < 0.001) while anti-inflammatory IL-10 production was increased by 45% (Fig. 1a; P < 0.01).

MSCs affect innate and adaptive immune responses.
(a) DCs were co-cultured with indicated ratios of MSCs for 3 hours prior to addition of 100 ng/mL LPS for 24 hours. DCs numbers remained constant. Cytokine responses were determined by ELISA. (b) Splenocytes from an LDLr KO mouse were co-cultured with indicated ratios of MSCs in the presence of αCD3/CD28 for 72 hours. Proliferation was assessed by 3H-thymidine incorporation and Ki-67 expression by flow cytometry. Splenocyte numbers remained constant. All values are expressed as mean ± SEM and representative of at least two independent experiments done in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001. ND defines not determined.
Since MSCs have been shown to affect T cell responses, we co-cultured MSCs with splenocytes from an atherosclerosis-prone LDLr KO mouse in the presence of αCD3/CD28 for 72 hours. MSCs potently inhibited T cell proliferation by 99%, as measured by 3H-thymidine incorporation, compared to αCD3/CD28 stimulated splenocytes in the absence of MSCs. CD4+ T cell proliferation was reduced by 92% and CD8+ T cell proliferation by 87%, as measured by Ki-67 expression. Polarization of all CD4+ T cell subsets was inhibited to a similar extent (Th1 and Th2 more than 99% and Tregs by 89%; Fig. 1b). Cytokine responses in the splenocyte co-culture indicated similar effects: IL-10 production was not detectable, IFN-γ was reduced by 99% and TNF-α showed a 92% reduction upon the co-incubation with MSCs. Co-cultures of isolated CD4+ T cells and MSCs showed comparable, but milder effects: proliferation of CD4+ T cells was on average reduced by 80%, the skewing towards specific T cell subsets was reduced by an average of 85% and cytokine production was also almost completely abolished (Supplementary Fig. 2). Interestingly, MSC culture supernatant had no effect on T cell proliferation, suggesting that cell-cell contact is crucial (Supplementary Fig. 3).
Before determining the effect of MSCs on atherosclerosis, we fluorescently labelled MSCs with CFSE and established the fate of these fluorescently labelled MSCs after intravenous injection into LDLr KO mice on a cholesterol rich diet (WTD) and determined to which organs they migrated (Supplementary Fig. 4). After fifteen minutes, MSCs accumulated primarily in the lungs and then slowly migrated out of the lungs. One to three hours after injection, we found that MSCs had migrated to the liver, the heart, the draining lymph nodes of the heart and the aorta. Surprisingly, only few MSCs were recovered in the spleen.
To test whether MSCs are able to modulate immune responses and thereby atherosclerosis, LDLr KO mice were treated with three intravenous injections of MSCs every other day prior to induction of atherosclerosis by WTD feeding. One day after the start of WTD, we found a significant 38% drop in circulating CD4+ T cells (control: 14.1% versus MSC: 8.7%, P < 0.05). Interestingly, we also observed an initial 51% increase in circulating Tregs (control: 9.2% versus MSC: 13.9%, P < 0.001; Fig. 2a). Eight weeks after inducing atherosclerosis, no difference in absolute white blood cell counts (data not shown) and percentage of circulating and splenic CD4+ T cells was found (Supplementary Fig. 5). A significant 18% decrease in splenic effector CD62L−/CD4+ T cells was observed (P < 0.001; Fig. 2b), while no effects on T cell subsets could be observed. In the circulation Th1 cells were significantly reduced by 44% (P < 0.01) and Tregs were reduced by 10% (P < 0.05). No significant effects of MSC therapy on Th2 cells were observed (Fig. 2b). Total CD8+ T cell numbers were not affected by MSC treatment in the circulation throughout the entire experiment (Fig. 3a) and were also not affected in the spleen after eight weeks of WTD (Supplementary Fig. 5). However, we again found a significant 25% decrease in effector CD62L−/CD8+ T cells in the circulation and spleen after eight weeks WTD upon MSC treatment (P < 0.05, Fig. 3b). To evaluate the proliferative capacity of T cells after MSC treatment, we isolated splenocytes eight weeks after induction of atherosclerosis by WTD feeding and cultured them in the presence of αCD3/CD28. In line with the in vitro data, splenocytes obtained from MSC-treated recipients showed a significant 30% decrease in T cell proliferation (P < 0.01, Fig. 3c). Furthermore, MSC treatment also significantly reduced circulating monocytes by 33% (P < 0.05), which was directly associated with a 77% reduction in serum CCL2 levels (P < 0.01; Fig. 3d), again suggesting a reduced inflammatory status of the MSC-treated mice. In line, although levels are just detectable, we observe a significant decrease in circulating IFN-γ both at 7 days (P < 0.01) and 8 weeks (P < 0.05) of WTD and a significant increase in circulating IL-10 levels (p < 0.05) in MSC treated animals after 8 weeks of WTD (Fig. 4a). Additionally, we observed a 20% reduction of IFN-γ (P = 0.05) and a 31% reduction of IL-6 (P < 0.05) in the draining lymph nodes of the heart. Moreover, we see a significant 76 % reduction of IFN-γ (P < 0.01) in the liver and a 33% reduction of TNF-α expression (P < 0.05) in white adipose tissue (Fig. 4b).

MSC treatment affects CD4+ T cell responses in vivo.
Male LDLr KO mice received three i.v. injections of either PBS (control) or 0.5 × 106 MSCs (MSC) and were then fed a Western-type diet (WTD) for eight weeks. (a) CD4+ T cells, as well as the percentage of FoxP3+ regulatory T cells (Treg) within CD4+ T cells, were measured in the circulation throughout the entire experiment by flow cytometry. (b) After eight weeks, effector CD4+ T cells, determined as CD62L− within CD4+ T cells, as well as T cell subsets of CD4+ T cells in the circulation and spleen were determined by flow cytometry. All values are expressed as mean ± SEM and representative of six mice per group. *P < 0.05, **P < 0.01, ***P < 0.001.

MSC treatment affects CD8+ T cell and monocyte responses in vivo.
(a) CD8+ T cells were measured in the circulation throughout the entire experiment by flow cytometry. (b) After eight weeks, effector CD8+ T cells, determined as CD62L− within CD8+ T cells, in the circulation and spleen were determined by flow cytometry. (c) Splenocytes were isolated and stimulated with αCD3/CD28 for 72 hours. Proliferation was assessed by the amount of 3H-thymidine incorporation. Proliferation is normalized for proliferation of controls (without stimulation) and expressed as the stimulation index (S.I.). (d) Circulating monocytes, determined as CD11bhiLy-6G−, were analyzed by flow cytometry. CCL2 levels in serum were determined by ELISA. All values are expressed as mean ± SEM and representative of six mice per group. *P < 0.05, **P < 0.01.

MSC treatment reduces pro-inflammatory cytokine response.
(a) Serum IL-6 levels were measured by ELISA, while IFNγ, TNFα and IL-10 levels were determined by luminex immunoassay. mRNA expression of IL-6, TNF-α, IFN-γ and IL-10 are shown for draining lymph nodes of the heart (hLN), liver and white adipose tissue (WAT). Expression is shown relative to the expression of three housekeeping genes (SDHA, HPRT and Rpl27). All values are expressed as mean ± SEM. Serum cytokine levels are representative of n = 3 pooled samples (7 days), n = 3 control and n = 6 MSC treated (8 weeks) and mRNA cytokine expression is representative of all mice. *P < 0.05. **P < 0.01.
As atherosclerosis is further determined by dyslipidaemia, we monitored weight and cholesterol levels throughout the entire experiment. Interestingly, although we did not observe a significant effect on weight or a direct effect of MSCs on plasma cholesterol levels after adoptive transfer, we found a significant 33% reduction in serum cholesterol levels in MSC-treated mice after eight weeks WTD (Fig. 5a). The decrease in cholesterol level was mainly a result of a decreased very low-density lipoprotein (VLDL) level as assessed by fast protein liquid chromatography analysis (P < 0.01; Fig. 5b). We found that the expression of hepatic lipoprotein lipase (LPL), which hydrolyses triglycerides into free fatty acids needed for VLDL synthesis, was significantly reduced by 34% (P < 0.01; Fig. 5c). In adult liver LPL is expressed by macrophages (Kupffer cells), which were not affected in their number as assessed by F4/80 and Clec4f expression, as well as CD68 expression (Supplementary Fig. 6). In white adipose tissue LPL regulates fatty acid uptake from triglyceride-rich lipoproteins for storage35. We found no difference in LPL expression in white adipose tissue (Supplementary Fig. 6), indicating that the reduction of LPL is specific for the liver.

MSC treatment reduces VLDL production.
(a) Body weight and cholesterol levels were monitored throughout the entire experiment. (b) Cholesterol distribution among plasma lipoprotein subclasses was determined by FPLC analysis after eight weeks WTD. For FPLC analysis serum of three mice was pooled. (c) Liver mRNA expression of lipoprotein lipase (LPL), (d) of SREBP-1c and SREBP-2 and (e) of acetyl-CoA carboxylase (Acc1), fatty acid synthase (FASN), stearoyl-CoA desaturase-1 (Scd1) is shown, relative to the expression of three housekeeping genes (SDHA, HPRT and Rpl27). All values are expressed as mean ± SEM and representative of all mice. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, we found a significant 27% reduction of Sterol regulatory element-binding protein (SREBP)-2 (P < 0.05) and a non-significant 18% reduction of SREBP-1c (P = 0.06; Fig. 5d); transcription factors activating multiple genes to promote triglyceride and cholesterol synthesis and thereby VLDL synthesis. Several genes involved in hepatic de novo lipogenesis, such as acetyl-CoA carboxylase 1 (ACC1) and fatty acid synthase (FASN) were not affected (Fig. 5e). Also diglyceride acyltransferase 1 (DGAT1), involved in the last steps of triglyceride synthesis and microsomal triglyceride transfer protein (MTTP), important for lipoprotein assembly, were not affected (Supplementary Fig. 6). However, strikingly, the rate-limiting enzyme in fatty acid synthesis stearoyl-CoA desaturase-1 (Scd1) was significantly reduced by 35% (P < 0.01; Fig. 5e). It should be noted that while expression levels are not necessarily identical to protein levels, these are predictive for VLDL metabolism and have been shown to correlate with atherosclerosis development36,37.
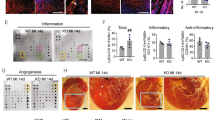
The beneficial effects of MSC treatment both on immune responses and cholesterol metabolism resulted in a significant 33% decrease in atherosclerotic lesion size in MSC-treated mice (1.4 × 105 ± 0.2 × 105 μm), compared with control mice (2.1 × 105 ± 0.3 × 105 μm, P < 0.05; Fig. 6a). Similar trends were observed when determining lesion area in all mice as percentage of vessel lumen, while lipid deposition was not affected (Supplementary Fig. 7). Additionally, we determined a significant 56% reduction in relative macrophage positive area of total lesion size (control: 29.4 ± 4.3% vs. MSC: 13.0 ± 2.6%, P < 0.01; Fig. 6b) and a 61% reduction of adventitial CD3+ T cells (control: 0.65 ± 0.12 cells per 105 μm2 vs. MSC: 0.25 ± 0.09 cells per 105 μm2, P < 0.01; Fig. 6c), indicating a reduced inflammatory status of the lesions. Lesion stability was not affected as the collagen content, determined by Masson’s trichrome staining, was not significantly different between the groups (control: 21.7 ± 1.1% vs. MSC: 19.4 ± 2.0%; Fig. 6d). Moreover, no difference in fibrous cap size and necrotic core size was observed (Supplementary Fig. 7), indicating that overall lesion composition was not affected.

MSC treatment reduces lesion development.
(a) Lesion size in the three valve area of the aortic root was determined; representative cross-sections stained with Oil-Red-O and hematoxylin are shown. (b) Macrophage content was determined by MOMA-2 staining as percentage of total lesion area. (c) Adventitial T cell numbers were determined by αCD3 staining. (d) Collagen content was determined by Masson’s Trichrome staining as percentage of total lesion area. Values are expressed as mean ± SEM and representative of all mice. *P < 0.05, **P < 0.01.
Discussion
Here we show that MSC treatment may be a promising strategy to reduce atherosclerotic lesion development. In agreement with previous studies1,16,18,19, we describe that MSCs can reduce the production of pro-inflammatory cytokines by DCs and dramatically inhibit T cell proliferation in vitro. We further show that adoptive transfer of MSCs into LDLr KO mice results in an overall reduced inflammatory state, as measured by reduced circulating IFN-γ and increased IL-10 levels. In addition, we observe reduced IL-6 expression in draining lymph nodes of the heart, reduced TNF-α expression in white adipose tissue and reduced IFN-γ expression in livers of MSC-treated mice. Moreover, MSC treatment results in an initial drop in circulating CD4+ T cell numbers. While this reduction was only observed initially after MSC transfer, a significant decrease in effector CD62L−/CD4+ and CD62L−/CD8+ T cells, as well as a significant decreased T cell proliferative capacity, was observed after eight weeks WTD in the spleen, clearly indicating a reduced differentiation of naïve T cells, which is consistent with effects observed in vitro. In agreement, a significant decrease in circulating Th1 cells and in Tregs was observed eight weeks after MSC transfer, as well as a significant reduction in adventitial CD3+ T cell numbers. However, directly after MSC treatment we observed an initial increase in circulating Tregs. This is in agreement with a recent study by Wang et al.38 who found that MSCs increased Tregs and thereby protect from atherosclerosis. However, in this study ApoE KO mice were treated multiple times with MSCs and with much higher numbers of MSCs (107) compared to our study (0.5 × 106), which ensured a longer increase of Tregs compared to our study. Since we observed an overall significant inhibition of T cell differentiation both in vivo and in vitro, we hypothesized that additional processes and cell types are needed for the observed increase in Tregs. It has been suggested that MSCs can induce apoptosis of T cells, which may explain the initial drop in CD4+ T cells that we observed in vivo and that these apoptotic T cells are cleared by phagocytes, which in turn may induce Tregs39. Therefore in vivo Tregs could initially be induced indirectly, while later the overall inhibition of T cell differentiation reduces Treg numbers. Additionally, MSC therapy significantly reduced circulating monocytes and serum CCL2 levels, again clearly suggesting reduced immune responses. This is in line with a previous study showing reduced monocytes after MSC therapy for myocardial infarction11. The reduced monocytes and their reduced recruitment to the lesions resulted in a dramatic 56% reduction in lesional macrophages.
Unexpectedly, we found significantly lower plasma cholesterol levels in MSC-treated mice, due to a reduction of VLDL levels, which is to our knowledge an effect that was not been previously described upon MSC treatment. The effects of MSC therapy on plasma cholesterol levels only emerged around four to five weeks after treatment indicating that the effects of MSCs could be rather indirect, e.g. by modulation of other cell types. Previous studies have found a connection between immune cells and cholesterol metabolism. For example lymphotoxins, which are expressed by CD4+ T cells and DCs, play a role in the homeostasis of these cells and can also contribute to metabolic disease40,41. CD4+ T cell expression of LIGHT, another ligand of the lymphotoxin receptor, increases plasma cholesterol levels, again showing a direct link between immune cells and cholesterol metabolism40. Furthermore, an increased lifespan of DCs42 and the adoptive transfer of mature DCs was found to correlate with decreased serum cholesterol levels43. Hence a modulation of immune cells by MSCs could also indirectly affect cholesterol metabolism. Interestingly, we find a significant reduction of LPL expression only in the liver and not in white adipose tissue. In the adult liver, LPL is exclusively expressed by the Kupffer cells44. As no difference in Kupffer cell markers was found, we can assume that LPL expression is specifically decreased by the Kupffer cells themselves. While LPL KO mice develop severe hypertriglyceridemia45,46, liver-specific LPL overexpression also results in hypertriglyceridemia47. Moreover, LPL deficiency in macrophages has been found to reduce their uptake of VLDL or oxLDL, thereby decreasing atherosclerosis48,49. Interestingly, TNF-α has been shown to induce LPL mRNA and activity in livers in several studies47, indicating that reduced LPL expression could be a direct result of reduced inflammation, as we see reduced TNF-α in white adipose tissue. However, it must me noted that, although levels are very low, we do not observe an effect of MSC treatment on circulating TNF-α levels. This suggests that indeed a modulation of macrophage function by MSC treatment could have occurred. Wang et al.38 described that foam cell formation is inhibited by MSCs, which suggests that MSC can indeed directly modulate macrophage function and activation in atherosclerosis. Interestingly, LPL is secreted and results in the breakdown of triglycerides into free fatty acids, which could result in less availability of free fatty acids for VLDL synthesis by hepatocytes. In addition, previous research has found that activated Kupffer cells can express mediators that promote VLDL secretion by hepatocytes50, indicating that a reduced activation of Kupffer cells in MSC-treated mice could result in reduced VLDL metabolism. However, also the reduced inflammatory environment could have directly affected VLDL synthesis. In line with this, TNF-α, which is downregulated upon MSC and splenocyte co-culture, has been shown to upregulate SREBP-1c51, thereby increasing VLDL synthesis. On the other hand, IL-10 overexpression has been shown to result in reduced plasma cholesterol, mostly due to reduced VLDL, in LDLr KO mice52. Overall, our data indicates that MSCs reduce VLDL levels by decreasing inflammation in treated mice and thereby affecting hepatocytes and Kupffer cells.
Overall MSC therapy was highly effective in reducing both immune responses and improving dyslipidemia, the two driving forces behind atherosclerosis and resulted in a significant 33% reduction in aortic root lesion sizes. Future studies will need to show the effect of MSCs on different stages of atherosclerosis, but our observations suggest that due to a combined effect on inflammation and plasma cholesterol levels a beneficial effect may be anticipated. For example, an increased stability of advanced lesions can be expected as IL-10 has been shown to promote lesion stability53,54. The fact that we do not observe an effect on lesion stability is directly associated with the early atherosclerotic lesions which contain a low amount of vascular smooth muscle cells. Recently, allogeneic MSCs were evaluated for their potential to repair ruptured lesions and were shown to increase regeneration of the inner endothelial lining and collagen fiber formation in the vessel wall55, implying their potential for the treatment of progressed lesions. Furthermore, a modulation of MSCs to enhance their anti-inflammatory capacities and/or their lifespan could prove interesting.
The drawback of some studies10,11 assessing the effect of MSCs on inflammation is that they employ human MSCs in SCID mice, which lack T and B cells. As these do not have a fully functional immune system it is difficult to examine the effect of MSCs on immune responses. Additionally, there have been reports that allogeneic MSCs can be rejected by recipient mice10, a caveat for a recent study by Lin et al.56. In this study human MSCs were transferred to ApoE KO mice, which increased eNOS in the aorta and thereby increased vasodilation in ApoE KO mice, but only resulted in a mild reduction of atherosclerosis. For these reasons, we performed an adoptive transfer of MHC-matched mouse MSCs into LDLr KO mice to avoid host immune responses to the grafted MSCs and to be able to do the experiment in mice with a functional immune system.
To translate our findings to clinical application, our studies could be in the future replicated using human MSCs in humanized mouse models for atherosclerosis, which can be humanized in their immune system, e.g. by transfer of human hematopoietic stem cells into SCID, NOG or NSG mice57,58. Also, a more humanized cholesterol metabolism, such as in the ApoE*3/CETP-Leiden mice59, would enable a confirmation of effects observed on VLDL metabolism. Taken together, our study provides evidence that a MSC-based treatment strategy has multifactorial therapeutic potential for inhibiting atherogenesis as it reduces inflammatory responses but also limits dyslipidaemia.
Materials and Methods
Animals
C57BL/6 and LDLr KO mice were originally obtained from the Jacksons Laboratory, kept under standard laboratory conditions and administered food and water ad libitum. All animal work was approved by the Ethics Committee for Animal Experiments of Leiden University and conforms to Dutch government guidelines.
DC culture
For DC cultures, bone marrow cells were isolated from the tibias and femurs of C57BL/6 mice. The cells were cultured for ten days at 37 °C and 5% CO2 in 10 cm2 petri dishes in IMDM supplemented with 10% FCS, 1% penicillin/streptomycin (all obtained from PAA), 1% glutamax (Thermo Fisher Scientific) and 20 μM β-mercaptoethanol (Sigma Aldrich) in the presence of 20 ng/mL granulocyte-macrophage colony-stimulating factor (Preprotech). DC purity was assessed by CD11c expression (flow cytometry) and routinely found to be above 95%.
MSC isolation, phenotyping and labelling
Bone marrow cells were obtained by flushing femurs with washing medium (RPMI supplemented with 2% FCS, penicillin, streptomycin and L-glutamine). Next the bone marrow cells were cultured in a 75 cm2 flask containing αMEM (Life Technologies) supplemented with 10% FCS (Greiner Bio-One), 2% penicillin/streptomycin (Life Technologies) and 1% L-Glutamine (Life Technologies; MSC medium). Subsequently, plastic adherent MSCs were cultured to 95% confluency in a fully humidified atmosphere at 37 °C and 5% CO2, harvested using trypsin and further expanded until sufficient numbers were obtained.
The MSCs that were used throughout this study were of passage six to eight; for in vivo MSC treatment cells of passage eight were used. MSC were phenotyped using the following antibodies: TER119 (clone TER119), CD31 (clone MEC 13.3), CD45.2 (clone 104), CD90.2 (clone 53-2.1), CD29 (clone HMb1-1), Sca-1 (Clone D7), CD105 (clone MJ7/18), CD44 (clone IM7) and CD106 (clone 429). All antibodies were obtained from BD Biosciences.
For tracking experiments, MSCs were labelled with 10 μM Carboxyfluorescein succinimidyl ester (CFSE) according to manufacturer’s protocol (Thermo Fisher Scientific). Briefly, MSCs were resuspended in prewarmed PBS/0.1% BSA at a concentration of 1 × 106 cells/mL and incubated with dye at 37 °C for 10 minutes in the dark. Afterwards, the staining was quenched by adding ice-cold media and incubating for another 5 minutes on ice, followed by three subsequent washing steps to remove excess CFSE.
Co-cultures
For DC-MSC co-cultures, 1 million DCs were plated in 2 cm2 non-tissue culture treated petri dishes with indicated ratios of MSCs for 24 hours in MSC medium. DCs were stimulated with 100 ng/mL LPS to determine cytokine responses. Three independent experiments were done in triplicate.
For splenocytes-MSC co-cultures, single cell suspensions of spleens from LDLr KO mice were obtained by using a 70 μm cell strainer (VWR International). Red blood cells were lysed with erythrocyte lysis buffer (0.15 M NH4Cl, 10 mM NaHCO3, 0.1 mM EDTA, pH 7.2). 2 × 105 splenocytes were added per well with indicated amounts of MSCs.
For T cell-MSC co-cultures, CD4+ T cells (>95% purity) were isolated from splenocytes by using the BD IMagTM mouse CD4 T lymphocyte enrichment set according to manufacturer’s protocol (BD Biosciences). 1 × 105 T cells were added per well with indicated amounts of MSCs.
Three independent experiments for both splenocytes and T cell-MSC co-cultures were cultured in quintuplicates in 96-well round-bottom plates in the presence or absence of αCD3/28 (2 μg/mL, eBioscience) for 72 hours in complete RPMI 1640, supplemented with 10% FCS, 100 U/ml penicillin/streptomycin, 2 mM L-glutamine (all obtained from PAA, Germany) and 20 μm β-mercaptoethanol (Sigma Aldrich). Proliferation was measured by Ki-67 expression by flow cytometry or addition of 3H-thymidine (0.5 μCi/well) for the last 16 hours of culture. The amount of 3H-thymidine incorporation was measured using a liquid scintillation analyzer (Tri-Carb 2900R) as the number of disintegrations per minute (dpm). T cell subsets were determined by flow cytometry.
We performed two independent experiments with splenocytes and T cell cultures in the presence of MSC culture supernatant in quintuplicates, MSC culture supernatant was added in different concentration of total medium added. MSC supernatant was filtered using a 0.2 μm filter to remove residual cells before use.
Atherosclerosis
Atherosclerosis was induced in 16 weeks old male LDLr KO mice by feeding a Western-type diet (WTD) (0.25% cholesterol and 15% cocoa butter; Special Diet Services). 12 Mice per group received 3 i.v. injections of PBS or 0.5 million MSCs every other day prior to eight weeks WTD.
Flow Cytometry
Mice were sacrificed and subsequently, blood and spleen were harvested of six mice per group. White blood cells were obtained as described above. 300,000 cells per sample were stained with the appropriate FACS antibodies. The following antibodies were used: CD11b-FITC (clone M1/70), CD11c-PE (clone N418), CD30L-PE (clone RM153), CD4-PerCP (clone RM4–5; BD Biosciences), CD40-FITC (clone HM40-3), CD8-PerCP (clone 53-6.7; BD Biosciences), CD80-PE (clone 16-10A1), CD86-APC (clone GL1), FoxP3-APC (clone FJK-16s), Gata-3-PE (clone TWAJ), Ki-67-FITC (clone SolA15), Ly-6G-APC (clone 1A8; BD Biosciences), MHCII-FITC (clone AF6-120.1), OX40L-PE (clone RM124L), PD-L2-APC (clone MIH18), RORγt-PE (clone AFKJS-9) and T-bet-Alexa Fluor 647 (clone eBio4B10). All antibodies were purchased from eBioscience, unless stated otherwise. For intracellular staining, cells were fixed and permeabilized according to the manufacturer’s protocol (eBioscience). FACS analysis was performed on the FACS Canto II and data were analyzed using FACS Diva software (BD Biosciences).
In Vivo Imaging Systems (IVIS)
For MSC tracking experiments, 1 × 106 MSCs were stained with CFSE and injected intravenously. Ex vivo imaging was performed by placing either whole animals or organs in the IVIS Lumina Imaging System (Xenogen) at indicated time points and analyzing fluorescence based on the manufacturer’s recommendations. Fluorescence intensity was quantified as photons/sec/cm2 by Living Image software (Xenogen).
Histological analysis
To determine plaque size, 10 μm cryosections of the aortic root of all mice were stained with Oil-Red-O and haematoxylin (Sigma Aldrich). Corresponding sections were stained for collagen fibers using the Masson’s Trichrome staining (Sigma Aldrich) or immunohistochemically with antibodies against a macrophage specific antigen (MOMA-2, polyclonal rat IgG2b, 1:1000, Serotec Ltd.). Goat anti-rat IgG alkaline phosphatase conjugate (dilution 1:100; Sigma Aldrich) was used as a secondary antibody and nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate as enzyme substrates. To determine the number of adventitial T cells, CD3 staining was performed using anti-mouse CD3 (clone SP7, 1:150, ThermoScientific). BrightVision anti-rabbit-HRP was used as secondary antibody (Immunologic). The section with the largest lesion and four flanking sections were analyzed for lesion size and collagen content, two flanking sections were analyzed for macrophage and T cell content. All images were analyzed using the Leica DM-RE microscope and LeicaQwin software (Leica Imaging Systems, UK). The percentage of collagen, macrophages and Oil-Red-O in the lesions was determined by dividing the collagen, MOMA-2- or Oil-Red-O positive area by the total lesion surface area. The plaque percentage was calculated by dividing plaque size by the total root area (i.e. plaque + lumen size). Relative cap area (in %) was calculated by measuring cap size as percentage of total plaque size.
Real-time PCR
mRNA was isolated from the liver, draining lymph nodes of the heart and white adipose tissue of all mice using the guanidium isothiocyanate method and reverse transcribed (RevertAid Moloney murine leukemia virus reverse transcriptase). Quantitative gene expression analysis was performed on a 7500 Fast real-time PCR system (Applied Biosystems) using SYBR Green technology. The expression was determined relative to the average expression of three housekeeping genes: succinate dehydrogenase complex, Subunit A (SDHA), hypoxanthine phosphoribosyltransferase (HPRT) and 60S ribosomal protein L27 (Rpl27). For used primer pairs please refer to Supplementary Table 1.
Cytokines
IL-10, TNF-α, IFN-γ in cell culture supernatant were determined by ELISA (BD Biosciences), while IL-10, TNF-α, IFN-γ serum levels were determined in 3–6 animals per group by luminex immunoassay (eBioscience, procartaplex) according to manufacturer’s protocol. CCL2 and IL-6 levels in serum were determined in all mice by ELISA (BD Biosciences) according to manufacturer’s protocol.
Serum cholesterol levels
During the experiment, mice were weighed and blood samples were obtained by tail vein bleeding. Serum concentrations of total cholesterol were determined by enzymatic colorimetric assays (Roche Diagnostics). Precipath (standardized serum; Roche Diagnostics) was used as internal standard. The distribution of cholesterol over the different lipoproteins in serum was determined by fractionation of 30 μl of serum using a Superose 6 column (3.2 × 300 mm, Smart-System; Pharmacia). Total cholesterol content of the effluent was determined as described above.
Statistical analysis
Values are expressed as mean ± SEM and were tested for normal distribution. Data of two groups were analyzed with a two-tailed Student’s T-test. Data of three or more samples were compared by one-way ANOVA and data of two groups with more than one variable were analyzed by two-way ANOVA, both followed by Bonferroni post-testing. Statistical analysis was performed using Prism (GraphPad). Probability values of P < 0.05 were considered significant.
Additional Information
How to cite this article: Frodermann, V. et al. Mesenchymal Stem Cells Reduce Murine Atherosclerosis Development. Sci. Rep. 5, 15559; doi: 10.1038/srep15559 (2015).
References
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–7 (1999).
Bernardo, M. E. & Fibbe, W. E. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell 13, 392–402 (2013).
Toma, C., Pittenger, M. F., Cahill, K. S., Byrne, B. J. & Kessler, P. D. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation 105, 93–8 (2002).
Mangi, A. A. et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat. Med. 9, 1195–201 (2003).
Atsma, D. E., Fibbe, W. E. & Rabelink, T. J. Opportunities and challenges for mesenchymal stem cell-mediated heart repair. Curr. Opin. Lipidol. 18, 645–9 (2007).
Barbash, I. M. et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration and body distribution. Circulation 108, 863–8 (2003).
Amado, L. C. et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc. Natl. Acad. Sci. USA 102, 11474–9 (2005).
Silva, G. V. et al. Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density and improve heart function in a canine chronic ischemia model. Circulation 111, 150–6 (2005).
Houtgraaf, J. H. et al. Intracoronary infusion of allogeneic mesenchymal precursor cells directly after experimental acute myocardial infarction reduces infarct size, abrogates adverse remodeling and improves cardiac function. Circ. Res. 113, 153–66 (2013).
Uccelli, A., Moretta, L. & Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 8, 726–36 (2008).
Dayan, V. et al. Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic Res. Cardiol. 106, 1299–310 (2011).
Németh, K. et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 15, 42–9 (2009).
Melief, S. M., Geutskens, S. B., Fibbe, W. E. & Roelofs, H. Multipotent stromal cells skew monocytes towards an anti-inflammatory interleukin-10-producing phenotype by production of interleukin-6. Haematologica 98, 888–95 (2013).
Melief, S. M. et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells 31, 1980–91 (2013).
Nauta, A. J., Kruisselbrink, A. B., Lurvink, E., Willemze, R. & Fibbe, W. E. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J. Immunol. 177, 2080–7 (2006).
Aggarwal, S. & Pittenger, M. F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105, 1815–22 (2005).
Beyth, S. et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood 105, 2214–9 (2005).
Bartholomew, A. et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp. Hematol. 30, 42–8 (2002).
Glennie, S., Soeiro, I., Dyson, P. J., Lam, E. W.-F. & Dazzi, F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 105, 2821–7 (2005).
Hansson, G. K. Inflammation, atherosclerosis and coronary artery disease. N. Engl. J. Med. 352, 1685–95 (2005).
Weber, C., Zernecke, A. & Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat. Rev. Immunol. 8, 802–15 (2008).
Taghavie-Moghadam, P. L., Butcher, M. J. & Galkina, E. V. The dynamic lives of macrophage and dendritic cell subsets in atherosclerosis. Ann. N. Y. Acad. Sci. 1319, 19–37 (2014).
Emeson, E. E., Shen, M. L., Bell, C. G. & Qureshi, A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am. J. Pathol. 149, 675–85 (1996).
Frostegård, J. et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 145, 33–43 (1999).
Zhou, X., Paulsson, G., Stemme, S. & Hansson, G. K. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J. Clin. Invest. 101, 1717–25 (1998).
Voloshyna, I., Littlefield, M. J. & Reiss, A. B. Atherosclerosis and interferon-γ: new insights and therapeutic targets. Trends Cardiovasc. Med. 24, 45–51 (2014).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–37 (2011).
Chistiakov, D. A., Sobenin, I. A., Orekhov, A. N. & Bobryshev, Y. V. Dendritic cells in atherosclerotic inflammation: the complexity of functions and the peculiarities of pathophysiological effects. Front. Physiol. 5, 196 (2014).
Foks, A. C., Lichtman, A. H. & Kuiper, J. Treating Atherosclerosis With Regulatory T Cells. Arterioscler. Thromb. Vasc. Biol. (2014), doi: 10.1161/ATVBAHA.114.303568.
Reinders, M. E. J. et al. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: results of a phase I study. Stem Cells Transl. Med. 2, 107–11 (2013).
Zappia, E. et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 106, 1755–61 (2005).
Augello, A., Tasso, R., Negrini, S. M., Cancedda, R. & Pennesi, G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum. 56, 1175–86 (2007).
Fan, H. et al. Pre-treatment with IL-1β enhances the efficacy of MSC transplantation in DSS-induced colitis. Cell. Mol. Immunol. 9, 473–81 (2012).
Le Blanc, K. et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet 371, 1579–86 (2008).
Weinstock, P. H. et al. Lipoprotein lipase controls fatty acid entry into adipose tissue, but fat mass is preserved by endogenous synthesis in mice deficient in adipose tissue lipoprotein lipase. Proc. Natl. Acad. Sci. USA 94, 10261–6 (1997).
Liu, X., Strable, M. S. & Ntambi, J. M. Stearoyl CoA desaturase 1: role in cellular inflammation and stress. Adv. Nutr. 2, 15–22 (2011).
Karasawa, T. et al. Sterol regulatory element-binding protein-1 determines plasma remnant lipoproteins and accelerates atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 31, 1788–95 (2011).
Wang, Z. X. et al. Mesenchymal stem cells alleviate atherosclerosis by elevating number and function of CD4+CD25+FOXP3+ regulatory T-cells and inhibiting macrophage foam cell formation. Mol. Cell. Biochem. 400, 163–172 (2014).
Akiyama, K. et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell 10, 544–55 (2012).
Lo, J. C. et al. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science 316, 285–8 (2007).
Upadhyay, V. & Fu, Y.-X. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat. Rev. Immunol. 13, 270–9 (2013).
Gautier, E. L. et al. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation 119, 2367–75 (2009).
Habets, K. L. L. et al. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice. Cardiovasc. Res. 85, 622–30 (2010).
Camps, L. et al. Lipoprotein lipase in lungs, spleen and liver: synthesis and distribution. J. Lipid Res. 32, 1877–88 (1991).
Coleman, T. et al. COOH-terminal disruption of lipoprotein lipase in mice is lethal in homozygotes, but heterozygotes have elevated triglycerides and impaired enzyme activity. J. Biol. Chem. 270, 12518–25 (1995).
Weinstock, P. H. et al. Severe hypertriglyceridemia, reduced high density lipoprotein and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J. Clin. Invest. 96, 2555–68 (1995).
Merkel, M. et al. Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J. Clin. Invest. 102, 893–901 (1998).
Babaev, V. R. et al. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J. Clin. Invest. 103, 1697–705 (1999).
Van Eck, M., Zimmermann, R., Groot, P. H., Zechner, R. & Van Berkel, T. J. Role of macrophage-derived lipoprotein lipase in lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 20, E53–62 (2000).
Bartolomé, N., Arteta, B., Martínez, M. J., Chico, Y. & Ochoa, B. Kupffer cell products and interleukin 1beta directly promote VLDL secretion and apoB mRNA up-regulation in rodent hepatocytes. Innate Immun. 14, 255–66 (2008).
Ruan, H. et al. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes 51, 3176–88 (2002).
Von Der Thüsen, J. H. et al. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in LDLr–/– mice. FASEB J. 15, 2730–2 (2001).
Mallat, Z. et al. Protective role of interleukin-10 in atherosclerosis. Circ. Res. 85, e17–24 (1999).
Potteaux, S. et al. Leukocyte-derived interleukin 10 is required for protection against atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 24, 1474–8 (2004).
Fang, S.-M. et al. Allogeneic bone marrow mesenchymal stem cells transplantation for stabilizing and repairing of atherosclerotic ruptured plaque. Thromb. Res. 131, e253–7 (2013).
Lin, Y.-L., Yet, S.-F., Hsu, Y.-T., Wang, G.-J. & Hung, S.-C. Mesenchymal Stem Cells Ameliorate Atherosclerotic Lesions via Restoring Endothelial Function. Stem Cells Transl. Med. 4, 44–55 (2014).
Ito, M. et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100, 3175–82 (2002).
Shultz, L. D. et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–89 (2005).
Van den Maagdenberg, A. M. et al. Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia. J. Biol. Chem. 268, 10540–5 (1993).
Acknowledgements
The authors would like to thank Edith Slot for technical assistance. This work was supported by the Dutch Heart Foundation (2009B093. 2012T083); and the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development and the Royal Netherlands Academy of Sciences, for the GENIUS project “Generating the best evidence-based pharmaceutical targets for atherosclerosis” (CVON2011-19).
Author information
Authors and Affiliations
Contributions
V.F. designed the study, performed and analyzed experiments and wrote the manuscript. J.v.D., M.v.P., P.v.S. and I.B. performed and analyzed experiments and provided intellectual input. J.K. and S.C.A.d.J. provided intellectual input and revised the manuscript. All authors reviewed the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Frodermann, V., van Duijn, J., van Pel, M. et al. Mesenchymal Stem Cells Reduce Murine Atherosclerosis Development. Sci Rep 5, 15559 (2015). https://doi.org/10.1038/srep15559
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15559
This article is cited by
-
Top Five Stories of the Cellular Landscape and Therapies of Atherosclerosis: Current Knowledge and Future Perspectives
Current Medical Science (2024)
-
Editorial Commentary: Top Five Stories of the Cellular Landscape and Therapies of Atherosclerosis: Current Knowledge and Future Perspectives
Current Medical Science (2024)
-
Autologous adipose mesenchymal stem cell administration in arteriosclerosis and potential for anti-aging application: a retrospective cohort study
Stem Cell Research & Therapy (2020)
-
Adipose-derived Mesenchymal Stromal Cells Modulate Lipid Metabolism and Lipid Droplet Biogenesis via AKT/mTOR –PPARγ Signalling in Macrophages
Scientific Reports (2019)
-
The differentiation of mesenchymal stem cells to vascular cells regulated by the HMGB1/RAGE axis: its application in cell therapy for transplant arteriosclerosis
Stem Cell Research & Therapy (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
