
Abstract
Cancer tissue sampling affects the identification of cancer characteristics. We aimed to clarify the source of differentially expressed genes (DEGs) in macro-dissected cancer tissue and develop a robust prognostic signature against the effects of tissue sampling. For estrogen receptor (ER)+ breast cancer patients, we identified DEGs in macro-dissected cancer tissues, malignant epithelial cells and stromal cells, defined as Macro-Dissected-DEGs, Epithelial-DEGs and Stromal-DEGs, respectively. Comparing Epithelial-DEGs to Stromal-DEGs (false discovery rate (FDR) < 10%), 86% of the overlapping genes exhibited consistent dysregulation (defined as Consistent-DEGs) and the other 14% of genes were dysregulated inconsistently (defined as Inconsistent-DEGs). The consistency score of dysregulation directions between Macro-Dissected-DEGs and Consistent-DEGs was 91% (P-value < 2.2 × 10−16, binomial test), whereas the score was only 52% between Macro-Dissected-DEGs and Inconsistent-DEGs (P-value = 0.9, binomial test). Among the gene ontology (GO) terms significantly enriched in Macro-Dissected-DEGs (FDR < 10%), 18 immune-related terms were enriched in Inconsistent-DEGs. DEGs associated with proliferation could reflect common changes of malignant epithelial and stromal cells; DEGs associated with immune functions are sensitive to the percentage of malignant epithelial cells in macro-dissected tissues. A prognostic signature which was insensitive to the cellular composition of macro-dissected tissues was developed and validated for ER+ breast patients.
Similar content being viewed by others

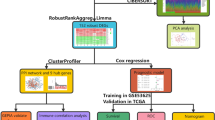

Introduction
Macro-dissected cancer tissues contain both carcinoma cells and stromal cells with distinct gene expression patterns1 and tissue sampling for gene expression profiling experiments commonly requires that the proportion of carcinoma cells is greater than certain threshold (e.g., 60%)2. However, because the proportions of carcinoma cells within distinct tumor locations of the same patient are quite different3, clinical sampling of macro-dissected cancer tissues could affect the identification of cancer characteristics, including differentially expressed genes (DEGs) and prognostic signatures.
To avoid this uncertainty, several deconvolution algorithms have been proposed to decompose gene expression profiles of macro-dissected samples into cell type-specific subprofiles4,5, but the requirement of the prior identification of signature genes of pure cells and the measurement of the proportion of cell types limits their application6. Another method to tackle this problem involves laser capture microdissection (LCM) technology to acquire a homogeneous collection of thousands of cells that are used to generate cell type-specific gene expression profiles7. For example, several researchers have identified DEGs of malignant epithelial cells and stromal cells and analyzed their roles in breast cancer progression8,9. LCM-coupled microarray studies typically use an additional round of RNA amplification (linear amplification) prior to microarray hybridization because LCM samples are generally too small to yield sufficient mRNA10,11,12,13. In some instances, RNA amplification introduces bias in the detection of gene expression values14,15. However, several studies have provided evidence of a clear correlation between signal intensities resulting from non-amplified mRNA compared with amplified mRNA16 with no substantial impact on the identification of DEGs between two groups of LCM samples in the same amplification step17.
In this study of estrogen receptor (ER)+ breast cancer patients, we identified DEGs for macro-dissected cancer tissues, malignant epithelial cells and stromal cells, defined as Macro-Dissected-DEGs, Epithelial-DEGs and Stromal-DEGs, respectively and compared them to reveal the cellular source of Macro-Dissected-DEGs. Then, we evaluated the correlation between expression measurements of DEGs identified in macro-dissected cancer tissues and the proportions of tumor cells in the tissues. Finally, we developed a prognostic signature based on the relative order of gene expression values that commonly occur in malignant epithelial cells and stromal cells compared with normal controls.
Results
Comparing Macro-Dissected-DEGs with Epithelial-DEGs and Stromal-DEGs
Using the Rankprod algorithm (see Methods), with 10% FDR control, we extracted DEGs in macro-dissected ER+ breast cancer tissues compared with normal controls from three datasets (M-Data1, M-Data2 and M-Data3, as described in Table 1), respectively. Pairwise comparisons of the three lists of DEGs showed that every two of the DEG lists were significantly overlapped (P-value < 1.0 × 10−12, hypergeometric test, see Methods the equation (1)). In addition, the dysregulation consistency scores of the overlapping DEGs of every two DEG lists, defined as the frequency of the overlapping DEGs that showed consistent up- or down-regulation in the two DEG lists, were 83–97%, which were all significantly higher than what expected by chance according to the binomial test (see Methods the equation (2), P-value < 2.2 × 10−16, Table S1). These results indicated that the DEGs identified in three independent datasets were significantly reproducible. We extracted DEGs that were dysregulated in the same directions in at least two of the three datasets to construct a list of DEGs that we defined as Macro-Dissected-DEGs.
Using the Rankprod algorithm18, with 10% FDR control, we identified two lists of DEGs in malignant epithelial cells compared with normal epithelial cells from two datasets of LCM samples for ER+ breast cancer (Lcm-Data1 and Lcm-Data2, as described in Table1), respectively. The two lists of DEGs contained 547 overlapping DEGs (P-value < 2.2 × 10−16, hypergeometric test), among which 97% were dysregulated in the same direction in the two lists. This result indicates that the DEGs of epithelial cells in two independent datasets were significantly reproducible (P-value < 2.2 × 10−16, binomial test, Table S1). Given that we could only identify a portion of DEGs in each dataset due to the small sample size19, we combined the two lists of DEGs of epithelial cells, deleted DEGs dysregulated in opposite directions and defined these genes as Epithelial-DEGs. For DEGs identified from the two LCM datasets from stromal cells, 77 DEGs overlapped between the two lists of DEGs (P-value < 2.2 × 10−16, hypergeometric test), among which 92% were dysregulated in the same direction (P-value < 2.2 × 10−16, binomial test, Table S1). Similarly, we integrated the two lists of DEGs of stromal cells, deleted DEGs dysregulated in opposite directions and defined these genes as Stromal-DEGs.
Among the 1251 overlapping genes between Epithelial-DEGs and Stromal-DEGs, 86.2% exhibited consistent dysregulation directions (defined as Consistent-DEGs) and the remaining 13.8% were dysregulated in opposite directions (defined as Inconsistent-DEGs). Then, we compared the Consistent-DEGs and Inconsistent-DEGs with Macro-Dissected-DEGs. The consistency score was 90.6% (P-value < 2.2 × 10−16, binomial test) among the 790 overlapping genes between Macro-Dissected-DEGs and Consistent-DEGs, which suggested that Consistent-DEGs for both epithelial and stromal cancer cells can be largely reflected in macro-dissected breast cancer tissue. In contrast, among the 91 overlapping genes between Macro-Dissected-DEGs and Inconsistent-DEGs, the consistency score was only 51.7% (P-value = 0.34, binomial test), which suggested that the differential expression signals of such Inconsistent-DEGs, when detected in macro-dissected tissues, were sensitive to the tissue compositions of epithelial and stromal cells. Obviously, the differential expression signals detected in macro-dissected tissues would be consistent with the epithelial DEGs only when the proportion of stromal cell is sufficiently small; otherwise, they would be affected by the stromal cells. Thus, when detected in macro-dissected tissues, the differential expression signals of these Inconsistent-DEGs would be different on datasets of macro-dissected tissues with different composition of epithelial and stromal cells and lack biological interpretation.
Functional interpretations of Macro-Dissected-DEGs
Based on the biological process (BP) of Gene Ontology (GO), using the GO-function algorithm20 designed for selecting non-redundant biologically relevant GO terms from GO terms significantly enriched with DEGs (see Methods), with FDR < 10%, we identified 238 GO terms that were significantly enriched with Macro-Dissected-DEGs. Among the 238 significant terms, 122 terms primarily involved in cell proliferation, developmental growth and cell division tended to be significantly enriched in Consistent-DEGs (P-value < 0.05, hypergeometric test, Table S2). This result suggested that cell proliferation and division processes observed in macro-dissected breast cancer tissue might reflect common alterations among malignant breast epithelial and surrounding stromal cells. Among the 238 significant terms, 18 terms primarily involved in immune responses, biological adhesion and the response to wounding tended to be significantly enriched in Inconsistent-DEGs (P-value < 0.05, hypergeometric test, Table S2). This result indicated that once these immune terms were enriched by Macro-Dissected-DEGs, other evidence was needed to reveal the source of the Macro-Dissected-DEGs.
The influence of cancer tissue composition on the prognostic signature
For the 376 gene expression profiles extracted from TCGA for ER+ breast cancer tissues which contained 60–100% tumor cell, we evaluated the correlation between the expression measurements of DEGs and the proportions of tumor cell by Pearson correlation analysis (see Methods). The results indicate that, when detected in macro-dissected tissues, the expression levels of 39.8% Consistent-DEGs and 47.8% Inconsistent-DEGs were significantly correlated with the proportions of tumor cell in the macro-dissected cancer tissues (P-value < 0.05, Pearson correlation). Thus, the measurement values of both Consistent-DEGs and Inconsistent-DEGs expression were sensitive to the tissue composition of epithelial and stromal cells.
We extracted the immune signatures developed by Nagalla et al.21 and Reyal et al.22 and compared the two lists of signatures with the DEGs identified in our study. The result indicate that some immune signatures were not dysregulated and others were oppositely deregulated in epithelial and stromal cells and these genes exhibit different dysregulated directions in macro-dissected breast cancer tissues (Table S3). These results demonstrated that immune-associated signatures were greatly affected by clinical cancer tissue sampling. Therefore, we developed a gene pair prognostic signature that was insensitive to the tissue composition of epithelial and stromal cells in macro-dissected breast cancer tissue.
Prognostic signature based on the relative order of expression
For Lcm-Data1, using the Fisher's exact test, with FDR < 10%, we extracted a list of gene pairs whose relative order of gene expression levels were significantly reversed in malignant epithelial cells compared with normal controls (see Methods). The similar process was performed for stromal cells. These two lists contained 56,268 overlapping gene pairs, among which 99.9% exhibited the same reversal patterns in malignant epithelial and stromal cells compared with normal controls, which was significantly more than expected by chance (P-value < 2.2 × 10−16, binomial test). We defined these gene pairs as Consistent-Gene-Pairs. When the Consistent-Gene-Pairs were compared with those extracted from Lcm-Data2, M-Data1, M-Data2 and M-Data1, the consistency scores were all greater than 99.70% (P-value < 2.2 × 10−16, binomial test, Table 2), suggesting that Consistent-Gene-Pairs were robust in different datasets.
Based on the integrated raining dataset (Sur-Data1 and Sur-Data2, as described in Table1) for macro-dissected ER+ breast cancer tissues with data of the relapse free survival (RFS), defined as the time period between the date of the first surgery and the date of first relapse, using the univariate Cox model with a FDR < 10%, we identified 17 gene pairs as prognostic gene pairs from the Consistent-Gene-Pairs. For each of the prognostic gene pairs presented in Table 3, the expression level of the latter gene was larger than that of the former gene in patients with better RFS and the orderings were reversed in patients with worse RFS.
According to the classification rule described in the Methods section, the prognostic gene pairs classified the training samples into a high-risk group with 53 samples and a low-risk group with 166 samples and the RFS of the high-risk patients was significantly reduced compared with low-risk patients (log-rank P = 4.15E–10, C-index = 0.66, Fig. 1). After adjusting for grade, age and tumor size using the multivariate Cox proportional hazards regression model, the prognostic gene pairs were identified as an independent prognostic signature for predicting patient outcomes (Table 4).

Kaplan–Meier curves illustrating relapse-free survival among patients with ER+ breast cancer based on Prognostic gene pairs in the training set.
The accuracy of the prognostic gene pairs was validated in two independent datasets. In Sur-Data3, the prognostic gene pairs classified the 209 patients into 101 high-risk patients and 108 low-risk patients and the RFS of the high-risk patients was significantly reduced compared with low-risk patients (log-rank P = 9.00E-04, C-index = 0.60, Fig. 2A). For Sur-Data4, disease-free survival (DFS) in 17 high-risk patients was significantly reduced compared with 102 low-risk patients classified by the prognostic gene pairs (log-rank P = 0.03, C-index = 0.57, Fig. 2B). In addition, the prognostic gene pairs were identified as an independent prognostic factor after adjusting for clinical factors, including grade, age and tumor size using the multivariate Cox proportional hazards regression model in the Sur-Data4 dataset, which contained additional clinical information (Table 4).

Kaplan–Meier curves illustrating relapse-free survival among patients with ER+ breast cancer based on Prognostic gene pairs in the test sets.
(A) Test set 1 consisted of Sur-Data3; (B) Test set 2 consisted of Sur-Data4.
Discussion
The impurity of macro-dissected cancer tissues raises several problems in the analyses of gene expression profiles in cancer tissues. In this study, we demonstrated that most DEGs related to proliferation and division processes observed in breast cancer macro-dissected tissues reflect similar gene expression changes in epithelial and stromal cells, whereas many immune DEGs observed in macro-dissected breast cancer tissues remain controversial. As opposed to epithelial cells, the dysregulation of surrounding stromal cells in breast cancer mainly includes immune-related functions, such as responses to wounds, immune responses and chemotaxis (Table S2 and Table S3). Given the distinct biological processes derived from epithelial and stromal cells, we should be cautious in interpreting DEGs identified from macro-dissected tissues and their related functions. We should also be cautious in interpreting immune related DEGs identified in macro-dissected tissues and micro-dissected stromal cells which include various types of cells, such as leukocytes, endothelial cells, fibroblasts, myofibroblasts and bone marrow-derived progenitors23.
Various studies have reported that genes associated with proliferation and immune responses could predict the outcomes of breast cancer patients22 and the expressional value of the immune gene prognostic signature is significantly associated with the relative abundance of tumor-infiltrating immune cells21. However, the clinical tissue sampling procedure is uncertain and our present analysis provides evidence that the expression measurements of these prognostic signatures tend to be influenced by the composition of the cancer tissue. To solve this problem, we developed a prognostic gene pairs index based on reversal of the relative order of gene expression values that commonly occur in malignant epithelial cells and stromal cells compared with their normal controls respectively, which is insensitive to the cellular composition of macro-dissected tissues. In addition, the rank-based predictors are more robust than absolute expression value-based predictors because they are rather robust against batch effects and insensitive to data normalization24. Furthermore, a rank-based predictor is feasible for individual-level prognostic analysis25.
In this study, we focused on breast cancer. It is likely that the same problem exists for other types of tumors; therefore, this subject requires further study.
Methods
Data sources and preprocessing
The ten gene expression datasets used in this study were downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/)26 and The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/)27. Three macro-dissected ER+ breast tissues with an average tumor cell proportion of approximately 60% were produced by different laboratories28,29,30 and two datasets for malignant epithelial cells and stromal cells of ER+ breast cancer and normal controls were produced by different laboratories8,9 (Table 1). These datasets were used to identify and compare Macro-Dissected-DEGs, Epithelial-DEGs and Stromal-DEGs. For the ER+ breast cancer data from TCGA31, the gene expression profile and the proportion of breast malignant epithelial cells were provided for each sample (Table 1) and these data were used to evaluate the correlation between expression values of DEGs identified in macro-dissected cancer tissues and the proportions of tumor cells in the tissues. Four datasets containing gene expression profiles32,33,34,35 and relapse-free survival (RFS) data of ER+ breast cancer patients with early-stage, lymph node negative (LN-) cancer who had not received adjuvant systemic treatment or hormone therapy were used to develop a prognostic signature (Table1).
For the GEO datasets, the raw data (.CEL files) from each dataset was processed using the Robust Multi-array Average (RMA) algorithm for background adjustment with quantile normalization36. Then, each probe-set ID was mapped to an Entrez gene ID with the custom CDF file. If multiple probe-sets were mapped to the same gene, the expression value for the gene was summarized as the arithmetic mean of the values of multiple probe-sets. Probe-set IDs with no mapped Entrez gene ID or Probe-set IDs that mapped to more than one Entrez gene ID were deleted. For the TCGA dataset, we applied the level 3 profile directly.
Identification of differentially expressed genes (DEGs)
The Bioconductor package RankProd18, based on the rank products algorithm37, was used to identify DEGs of breast cancer versus normal control samples with a false discovery rate (FDR) less than 10%. The P-values were adjusted using the Benjamini-Hochberg procedure38. A DEG was considered upregulated (or downregulated) if its average expression level in the cancer samples was increased (or reduced) compared with normal controls.
Evaluation of the consistency of two DEG lists
If DEG list 1 with L1 genes and DEG list 2 with L2 genes have k overlapping genes, the probability (P1) of observing at least k overlapping genes by chance can be calculated according to the following cumulative hypergeometric distribution model:

where L represents the number of the background genes commonly detected in the datasets from which the DEGs are extracted. The two DEG lists were considered to be significantly overlapping if P1 < 0.05.
If a DEG exhibited the same dysregulated direction (up- or down-regulated) in the two DEG lists, it was considered consistent across the datasets. We defined a dysregulation consistency score as the percentage of consistent DEGs in the overlapping DEGs between the two DEG lists. The probability (P2) of observing at least s DEGs with the same dysregulation direction across the two datasets from k randomly selected genes was calculated according to the following cumulative binomial distribution model39:

where p represents the random possibility (here 0.5) of one DEG having the same dysregulated direction across two DEGs lists. A dysregulation consistency score was considered significant if P2 < 0.05.
Functional enrichment analysis
To derive biologically relevant, non-redundant terms from statistically significant terms for a disease, GO-function20 was used to select the disturbed functional categories significantly enriched in DEGs. We focused on analyzing the biological process (BP) of Gene Ontology (GO), which was downloaded in April 2013.
Correlation between the expression measurements of DEGs and the proportions of tumor cell in macro-dissected tissues
Extracted from TCGA, the 376 gene expression profiles for ER+ breast cancer tissues included the data of tumor cell proportions. Using these samples, for the Consistent-DEGs and Inconsistent-DEGs respectively, we applied Pearson’s correlation analysis to detect genes whose expression levels were significantly correlated with tumor cell proportions. Then, the percentages of DEGs that were significantly correlated with tumor cell proportions were calculated for Consistent-DEGs and Inconsistent-DEGs, respectively.
Development of the prognostic signature based on reversed gene pairs
For a pair of genes, gene A and gene B, we used Fisher’s exact test to evaluate whether the frequency of samples with a higher (or lower) expression level of gene A than gene B in disease samples was significantly different from that in the corresponding normal controls. The P-values were adjusted using the the Benjamini-Hochberg procedure38. The significant gene pairs detected with a FDR control level of 10% were defined as significantly reversed gene pairs. Gene pairs with the same reversals of relative ordering of gene expression measurements in malignant epithelial cells and stromal cells were defined as Consistent-Gene-Pairs.
Then, based on the expression profiles of ER+ breast cancer with RFS information, a univariate Cox regression model was used to select gene pairs among the Consistent-Gene-Pairs with a relative order of expression that was significantly correlated with the RFS; these pairs were defined as prognostic gene pairs. The prognostic classifier was constructed according to the following rule: a patient was classified into the low risk group if there were significantly more prognostic gene pairs classifying her as low risk (P-value < 0.05, binomial test); otherwise, the patient was classified into the high risk group. The multivariate Cox proportional hazards regression model was performed to determine whether prognostic gene pairs are an independent prognostic factor in predicting RFS after adjusting for clinical factors, such as age, grade and tumor size.
All statistical analyses were performed using the R 2.15.3 (http://www.r-project.org/).
Additional Information
How to cite this article: Xu, H. et al. The influence of cancer tissue sampling on the identification of cancer characteristics. Sci. Rep. 5, 15474; doi: 10.1038/srep15474 (2015).
References
Clarke, J., Seo, P. & Clarke, B. Statistical expression deconvolution from mixed tissue samples. Bioinformatics 26, 1043–1049 (2010).
West, M. et al. Predicting the clinical status of human breast cancer by using gene expression profiles. P Natl Acad Sci Usa 98, 11462–11467 (2001).
Angell, H. & Galon, J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol 25, 261–267 (2013).
Ghosh, D. Mixture models for assessing differential expression in complex tissues using microarray data. Bioinformatics 20, 1663–1669 (2004).
Erkkila, T. et al. Probabilistic analysis of gene expression measurements from heterogeneous tissues. Bioinformatics 26, 2571–2577 (2010).
Zhao, Y. & Simon, R. Gene expression deconvolution in clinical samples. Genome Med 2, 93 (2010).
Espina, V., Milia, J., Wu, G., Cowherd, S. & Liotta, L. A. Laser capture microdissection. Mimb 319, 213–229 (2006).
Ma, X. J., Dahiya, S., Richardson, E., Erlander, M. & Sgroi, D. C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res: Bcr 11, R7 (2009).
Casey, T. et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Tr 114, 47–62 (2009).
Kube, D. M. et al. Optimization of laser capture microdissection and RNA amplification for gene expression profiling of prostate cancer. Bmc Mol Biol 8, 25 (2007).
Upson, J. J. et al. Optimized procedures for microarray analysis of histological specimens processed by laser capture microdissection. J Cell Physiol 201, 366–373 (2004).
King, C. et al. Reliability and reproducibility of gene expression measurements using amplified RNA from laser-microdissected primary breast tissue with oligonucleotide arrays. J Mol Diagn: Jmd 7, 57–64 (2005).
van Haaften, R. I. et al. Biologically relevant effects of mRNA amplification on gene expression profiles. Bmc Bioinformatics 7, 200 (2006).
de Bruin, E. C. et al. Macrodissection versus microdissection of rectal carcinoma: minor influence of stroma cells to tumor cell gene expression profiles. Bmc Genomics 6, 142 (2005).
Michel, C. et al. Liver gene expression profiles of rats treated with clofibric acid: comparison of whole liver and laser capture microdissected liver. Am J Pathol 163, 2191–2199 (2003).
Schneider, J. et al. Systematic analysis of T7 RNA polymerase based in vitro linear RNA amplification for use in microarray experiments. Bmc Genomics 5, 29 (2004).
Klee, E. W. et al. Impact of sample acquisition and linear amplification on gene expression profiling of lung adenocarcinoma: laser capture micro-dissection cell-sampling versus bulk tissue-sampling. Bmc Med Genomics 2, 13 (2009).
Hong, F. et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22, 2825–2827 (2006).
Zhang, M. et al. Apparently low reproducibility of true differential expression discoveries in microarray studies. Bioinformatics 24, 2057–2063 (2008).
Wang, J. et al. GO-function: deriving biologically relevant functions from statistically significant functions. Brief Bioinform 13, 216–227 (2012).
Nagalla, S. et al. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol 14, R34 (2013).
Reyal, F. et al. A comprehensive analysis of prognostic signatures reveals the high predictive capacity of the proliferation, immune response and RNA splicing modules in breast cancer. Breast Cancer Res: Bcr 10, R93 (2008).
Horimoto, Y., Polanska, U. M., Takahashi, Y. & Orimo, A. Emerging roles of the tumor-associated stroma in promoting tumor metastasis. Cell Adhes Migr 6, 193–202 (2012).
Zhou, X. et al. A relative ordering-based predictor for tamoxifen-treated estrogen receptor-positive breast cancer patients: multi-laboratory cohort validation. Breast Cancer Res Tr 142, 505–514 (2013).
Wang, H. et al. Individual-level analysis of differential expression of genes and pathways for personalized medicine. Bioinformatics 31, 62–68 (2015).
Barrett, T. et al. NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Res 35, D760–765 (2007).
Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. NATURE 490, 61–70 (2012).
Chen, D. T. et al. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res Tr 119, 335–346 (2010).
Pedraza, V. et al. Gene expression signatures in breast cancer distinguish phenotype characteristics, histologic subtypes and tumor invasiveness. Cancer 116, 486–496 (2010).
Clarke, C. et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis 34, 2300–2308 (2013).
Michailidou, K. et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45, 353–361, 361e351-352 (2013).
Desmedt, C. et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res: an official journal of the American Association for Cancer Research 13, 3207–3214 (2007).
Loi, S. et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. P Natl Acad Sci Usa 107, 10208–10213 (2010).
Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365, 671–679 (2005).
Ivshina, A. V. et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res 66, 10292–10301 (2006).
Irizarry, R. A. et al. Exploration, normalization and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Breitling, R., Armengaud, P., Amtmann, A. & Herzyk, P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. Febs Lett 573, 83–92 (2004).
Hochberg, Y. B. A. Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 57, 289–300 (1995).
Bahn, A. K. Application of binomial distribution to medicine: comparison of one sample proportion to an expected proportion (for small samples). Evaluation of a new treatment. Evaluation of a risk factor. Am J Med Genet A 24, 957–966 (1969).
Acknowledgements
This work was supported by the National Natural Science Foundation of China [Grant No. 81372213 and Grant No. 81201822]; and the Research Fund for the Doctoral Program of Higher Education of China [Grant No. 20112307110011]. The funders had no role in study design, data collection and analysis, preparation of the manuscript or decision to publish.
Author information
Authors and Affiliations
Contributions
All authors meet the authorship requirements. G.Z. and H.X. conceived and designed the analysis. H.X., X.G. and Q.S., collected the macro-dissected datasets and L.C.M. datasets and performed all the data analyses. Y.L., L.B.C. and M.M.Z. collected the survival datasets and downloaded the biological process (BP) of Gene Ontology (GO). H.X. drafted the manuscript. Z.G., W.Y.Z., H.X., L.S.Q. and Y.Y.G. revised the manuscript critically for important intellectual content. W.Y.Z., Z.G. and H.X. were agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xu, H., Guo, X., Sun, Q. et al. The influence of cancer tissue sampling on the identification of cancer characteristics. Sci Rep 5, 15474 (2015). https://doi.org/10.1038/srep15474
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15474
This article is cited by
-
Identification of EMT-related high-risk stage II colorectal cancer and characterisation of metastasis-related genes
British Journal of Cancer (2020)
-
Identification and characterization of genes with absolute mRNA abundances changes in tumor cells with varied transcriptome sizes
BMC Genomics (2019)
-
Robust transcriptional signatures for low-input RNA samples based on relative expression orderings
BMC Genomics (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
