
Abstract
We report the experimental observation and theoretical explanation of an unconventional interplay between divalent Co and trivalent Y dopants, both of which incur oxygen vacancies in the CeO2 host that has predominantly tetravalent Ce cations. The Co dopant atoms were experimentally found to act as a switch that turns on the dormant effect of Y-modulated band-gap reduction. As revealed by density functional theory (DFT) calculations with structures verified by synchrotron-radiation x-ray measurements, a Co 3d band that hybridizes with Ce 4f band was lowered due to reduced O 2p repulsion arising from oxygen vacancies incurred by Y doping and therefore gave rise to the observed band-gap narrowing effect. Such switch-and-modulator scheme for band-gap engineering in nanocrystal materials can lead to important applications in environmental protection and solar energy harvesting technologies.
Similar content being viewed by others

Introduction
Controllable modulation of energy band gaps in crystalline materials can lead to novel applications in a wide variety of technologies ranging from catalysis, photovoltaics to optical and electronic devices. We have attempted to modulate the band gap of CeO2 (ceria) nanocrystals by incorporating heterovalent dopant elements into the material. Nanostructures of ceria are widely used in the development of solar cells and environmental protection catalysts1,2,3. Oxygen vacancies in the form of minority trivalent cerium in CeO2, where Ce are predominantly tetravalent, play an important role in affecting the electronic structures and magnetism of ceria4,5,6. The catalytic activity of nanostructured CeO2 catalysts has also been found to increase substantially with the oxygen vacancy concentration due to vacancy-induced electronic structural changes2. Therefore, the electronic properties of CeO2 may presumably be manipulated by defect engineering techniques that alter the oxygen vacancy concentration, such as doping impurity atoms with valences less than 4+. In fact, a previous report has shown that doping of trivalent Nd can indeed lead to non-monotonic band-gap changes in ceria nanoparticles7.
In this work, we chemically incorporated trivalent Y and divalent Co dopant ions into ceria nanocrystals attempting to modulate the band gap of the doped host. Presumably, the only major effect of divalent Co ions in trivalent-Y-ion-doped ceria may simply be generating more oxygen vacancies and therefore enhance the existing effects incurred by yttrium. However, contrary to the above reasoning, our experimental measurements have shown that the divalent Co dopant atoms actually play a much more subtle role, namely a switch that turns on the dormant Y-modulated band-gap variation in Y-doped CeO2. To understand the mechanism of such unconventional interplay between Y and Co dopants, the crystal structure of the nanocrystal CeO2 host and the local structures surrounding the Y and Co dopant atoms were precisely determined by synchrotron radiation x-ray techniques. First-principle theoretical calculations were then carried out to obtain electronic structures of the codoped nanocrystal system with initial guess of structural model suggested by the x-ray analyses.
A polyol method was used to prepare the samples of Y-doped and (Y, Co)-codoped CeO2 nanocrystals. A slurry of Y(NO3)3.6H2O, CoCl2.6H2O and Ce(NO3)3.6H2O in diethylene glycol was vigorously stirred and heated to 160 °C. After 1 ml NH4OH was injected, the solution was continually heated for 120 minutes at 160 °C under reflux. The resulting reacting mixture was cooled to room temperature to form a suspension. Finally, a yellow precipitate was collected from the suspension by centrifugation, repeatedly washed with a mixture of deionized water and ethanol and then dried in an oven to form a powder sample. The Y and Co dopant concentrations were determined by inductively coupled plasma mass spectrometry (ICPMS). For the (Y, Co)-codoped samples, A1, A2, A3, A4 and A5, the Y concentrations are 0, 3.6, 7.1, 13.4 and 18.7 at.%, respectively, while the Co concentrations are about 4 at.%. For samples doped with Y only, B1, B2, B3, B4 and B5, the Y concentrations are 0, 3.3, 6.3, 12.2 and 16.7 at.%, respectively.
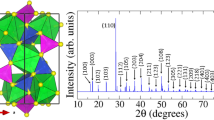
To determine the long-range-order crystal structures, x-ray powder diffraction (XRD) measurements were carried out for all samples using synchrotron radiation. As shown in Fig. 1, the CeO2 (111), (200), (220), (311), (222) and (400) Bragg peaks appear in the XRD patterns indicating cubic CeO2 structure for all samples. The lattice constant of the dopant-free sample 5.4160 Å is close to the bulk-ceria value 5.4111 Å. Due to the much smaller ion radius of Co than that of Ce, the lattice constant of the sample doped with Co only is reduced to 5.4001 Å. When Y is incorporated into the samples, the lattice constant decreases monotonically with increasing Y concentration from 5.4001 Å and 5.4160 Å for samples A1 and B1 to 5.3920 Å and 5.4114 Å for samples A5 and B5 in the Y-doped and (Y, Co)-codoped series, respectively. Such Y-induced lattice contraction may arise from the increase of oxygen vacancy concentration due to Y doping as demonstrated in previous reports3,8. The crystallite sizes were determined from XRD by using the Scherrer equation9 to be around 4–5 nm for all samples.

X-ray powder diffraction patterns and lattice parameter vs. Y concentration curves for the Y doped (upper) and the (Y, Co) codoped (lower) samples.
Curves have been shifted vertically for the sake of clarity.
Local structures surrounding Y and Co dopant atoms in the CeO2 host were probed by using the extended x-ray absorption fine structure (EXAFS) technique at beamlines BL07A and BL17C of National Synchrotron Radiation Research Center (NSRRC) in Taiwan. Conventional fluorescence mode was adopted using a Lytle fluorescence detector for Y K-edge and Co K-edge absorption measurements10,11. An established data reduction method was used to extract χ functions from the raw experimental data12. The Fourier transforms and EXAFS χ functions for the Y K-edge and Co K-edge EXAFS are plotted as fine lines in Figs 2 and 3, respectively. Local structural parameters were extracted using a curve-fitting procedure based on the FEFF software10,13. The amplitude reduction factor S02 representing the central atom shake-up and shake-off effects and the mean free path of photoelectrons λ were set to be 0.9 and 10 Å for the Y data and 0.68 and 10 Å for the Co data as determined from curve-fitting the EXAFS data for Y2O3 and CoO model compounds, respectively11,14. The curve-fittings exhibit a O shell at 2.31 ± 0.01 Å and a Ce shell at 3.77 ± 0.01 Å from the Y central atom. In comparison to the Ce-O distance of 2.34 Å and Ce-Ce distance of 3.83 Å of CeO2, the observed Y-O and Y-Ce distances indicate that the Y dopant atoms most likely occupy Ce sites in the CeO2 matrix. However, the coordination number of the nearest O shell surrounding Y is around 6.0 ± 0.5 to 7.0 ± 0.6, roughly one less than that of 8 surrounding Ce in the CeO2 host. The reduced coordination number of the nearest O shell indicates that an oxygen vacancy is generated around the Y dopant atom. The Co EXAFS data show only the first O shell at distances around 2.05 ± 0.01–2.07 ± 0.01 Å surrounding the Co atoms, which is very different from the Ce-O bond length. The coordination number of the nearest O shell surrounding Co is around 5.2 ± 0.4 to 5.8 ± 0.1, roughly two less than that surrounding Ce. This indicates that Co atoms may occupy Ce sites and are surrounded by around two oxygen vacancies that give rise to large distortion. As a consequence of the large distortion, short-range order was not observed by EXAFS beyond the nearest neighboring shell.

(a) Y K-edge EXAFS data for (Y, Co) codoped CeO2 samples. (b) Y K-edge EXAFS data for Y doped CeO2 samples. Fine lines: experimental; Coarse lines: curve fitting. Curves have been shifted vertically for the sake of clarity.

Co K-edge EXAFS data for (Y, Co) codoped CeO2 samples.
To determine the effective valencies of Y and Co in the samples, the Y and Co K-edge near-edge x-ray absorption fine structure (XANES) spectra for the doped ceria samples are plotted with those of model compounds of different Y or Co valencies in Fig. 4. As demonstrated in the insets, the XANES spectra for all samples are very similar to each other at both the Y and Co K edges indicating similar local bonding environment surrounding Y and Co in all samples, respectively. By comparing the edge position of the samples with those of model compounds, it is clear that the effective valencies for Y and Co in the ceria samples are 3+ and 2+, respectively. Therefore, it is confirmed by the XANES data that trivalent Y and divalent Co dopants were incorporated in the CeO2 host of tetravalent Ce.

(a) Y K-edge XANES data for Y doped and (Y, Co) codoped CeO2 samples. (b) Co K-edge XANES data for (Y, Co) codoped CeO2 samples.
The band gap variation due to Y and Co doping was monitored by UV-vis diffuse reflectance spectra as shown in Fig. 5. The band gap energies, associated with O 2p to Ce 4f transition, are determined by fitting the absorption data with Kubelka-Munk function15 and Tauc’s plots16 to be 2.74 eV, 2.58 eV, 2.43 eV, 2.31 eV and 2.28 eV, for the (Y, Co) codoped CeO2 samples, A1, A2, A3, A4 and A5, respectively. The band gap energies for samples doped with Y only, B1, B2, B3, B4 and B5, are 2.80 eV, 2.83 eV, 2.81 eV, 2.84 eV and 2.80 eV, respectively. The band gap of Co-free Y-doped CeO2 appears to remain largely unchanged as the Y concentration increases from 0 to 16.7%. However, when Co codopant of concentration 4% is added to the Y-doped CeO2, the band gap decreases monotonically from 2.74 eV to 2.28 eV as the Y concentration increases from 0 to 18.7%. As shown in the inset of Fig. 5, the Co and Y dopants act as a switch and a modulator for band gap tuning, respectively. It is worth noting that, as the band gap decreases with increasing Y concentration in the (Y, Co)-codoped samples, the lattice parameter also decreases monotonically as indicated by the XRD data (Fig. 1). Therefore, the possible mechanism adopted by previous reports that lattice relaxation leads to band-gap reduction is not applicable to our case7,17. As a matter of fact, the increase of Y concentration, while increasing oxygen vacancy concentration according to our EXAFS analysis, does not substantially reduce the band gap in the absence of the Co codopant.

(a) UV–vis diffuse reflectance spectra for the Y doped (lower) and the (Y, Co) codoped (upper) samples. Inset: A plot of band gap energy vs. Y concentration.
To understand the observed switch-and-modulator effect, first-principle electronic structures calculations based on the density functional theory (DFT) were performed using the Vienna ab initio simulation package (VASP)18 with an initial guess of structural model suggested by the x-ray analyses. In our calculations, a generalized gradient approximation (GGA) PBEsol functional19 was used. Compared to other methods, the GGA PBEsol+U method can better reproduce structural parameters and therefore are more suitable for our purpose20,21,22. A Hubbard U correction (GGA+U) to the Ce 4f orbitals with an effective U of 6.5 eV was employed to describe the correlation effects of the Ce 4f states in defective CeO2 crystals. A 2 × 2 × 2 supercell of the cubic Fm m fluorite structure with 96 atoms therein was used as the initial model for pure CeO2. The Brillouin-zone (BZ) integration was performed using a 3 × 3 × 3 Monkhorst- Pack k-point grid23. The plane-wave kinetic energy cutoff was set to be 400 eV20. Atomic positions and lattice parameters of all models were optimized without any symmetry restrictions until the maximum force on each atom was smaller than 0.025 eV/Å. To mimic oxygen vacancies, O atoms were removed from the pure CeO2 supercell. For the sample doped with Y, a Ce atom next to an oxygen vacancy was replaced by a Y atom. For the sample doped with Co, a Co atom substitutes for a Ce atom, where two O neighboring atoms are removed from the diagonal sites of a CoO8 cube. Finally, the (Y, Co)-codoped CeO2 model was constructed with Co at the center and Y on the edges of the supercell (Fig. 6). The dependence of band gap on Y concentration was investigated by comparing the result of one Y atom per supercell (3.1 at.%) to that of two Y atoms per supercell (6.2 at.%). The concentration of Co was one Co atom per supercell (3.1 at.%). After structural relaxation and stabilization, the calculated lattice constant 5.3929–5.4107 Å are close to the experimental values 5.3920–5.4160 Å. The calculated average Y-O and Co-O bond lengths are 2.298 Å and 2.00 Å, which are close to the experimental values of 2.31 ± 0.01 Å and 2.05 ± 0.01–2.07 ± 0.01 Å from EXAFS analysis, respectively. Since the calculated structures agree well with the x-ray results, the calculated densities of states (Fig. 7) are expected to be highly reliable.
m fluorite structure with 96 atoms therein was used as the initial model for pure CeO2. The Brillouin-zone (BZ) integration was performed using a 3 × 3 × 3 Monkhorst- Pack k-point grid23. The plane-wave kinetic energy cutoff was set to be 400 eV20. Atomic positions and lattice parameters of all models were optimized without any symmetry restrictions until the maximum force on each atom was smaller than 0.025 eV/Å. To mimic oxygen vacancies, O atoms were removed from the pure CeO2 supercell. For the sample doped with Y, a Ce atom next to an oxygen vacancy was replaced by a Y atom. For the sample doped with Co, a Co atom substitutes for a Ce atom, where two O neighboring atoms are removed from the diagonal sites of a CoO8 cube. Finally, the (Y, Co)-codoped CeO2 model was constructed with Co at the center and Y on the edges of the supercell (Fig. 6). The dependence of band gap on Y concentration was investigated by comparing the result of one Y atom per supercell (3.1 at.%) to that of two Y atoms per supercell (6.2 at.%). The concentration of Co was one Co atom per supercell (3.1 at.%). After structural relaxation and stabilization, the calculated lattice constant 5.3929–5.4107 Å are close to the experimental values 5.3920–5.4160 Å. The calculated average Y-O and Co-O bond lengths are 2.298 Å and 2.00 Å, which are close to the experimental values of 2.31 ± 0.01 Å and 2.05 ± 0.01–2.07 ± 0.01 Å from EXAFS analysis, respectively. Since the calculated structures agree well with the x-ray results, the calculated densities of states (Fig. 7) are expected to be highly reliable.

Schematic view of (Y, Co) codoped CeO2 structural models.
The large red balls, large green balls, large yellow balls and small blue balls represent the O, Ce, Y and Co atoms, respectively.

Total and partial densities of states for (a) Pure CeO2, (b) oxygen vacancy (VO) doped CeO2, (c) Y-doped CeO2, (d) Co-doped CeO2 and (e) (Y, Co)-codoped CeO2 with 3.1 at.% of Y and (f) (Y, Co)-codoped CeO2 with 6.2 at.% of Y. To clearly compare the band gaps, O 2p band edge of all structures were aligned in the plot.
The calculated O 2p to Ce 4f band gap for pure CeO2 (2.53 eV) is ~0.3 eV smaller than the experimental value of 2.80 eV. This is expected since GGA is well-known to underestimate band gap values. When an oxygen vacancy is introduced, a peak representing the f states localized around the two Ce ions nearest to the O vacancy appears in the middle of the band gap without changing the gap value (Fig. 7b). Doping with Y alone does not change the general shape of the density of states (Fig. 7c). Doping with Co alone can introduce two Co 3d impurity bands; one is in the middle of the band gap and the other hybridizes with Ce 4f orbitals right below the conduction band edge (Fig. 7d). While the O 2p to Ce 4f band gap remains unchanged if only one of the two dopants is incorporated, substantial band gap narrowing occurs when both Y and Co are present. At the Y concentrations of 3.1 at.% and 6.2 at.%, the calculated band gap is reduced from 2.53 eV of the pure ceria to 2.23 eV (Fig. 7e) and 2.17 eV (Fig. 7f), respectively. In comparison, the experimental band gap dropped from 2.74 eV to 2.58 eV and 2.43 eV as Y concentration increases from 0 at.% to 3.6 at.% and 7.1 at.%, respectively. Except for the underestimated band gap values of the GGA method, our calculations well reproduced the general trend of the Co-triggered and Y-modulated band gap variations observed experimentally. From the partial density of states plotted in Fig. 7, it is obvious that the Co 3d band right below the Ce 4f band edge plays a pivotal role in the observed Y-modulated band gap narrowing effect. As the Y concentration is increased, more oxygen vacancies are generated. Therefore, the repulsion of O 2p on Co 3d is lowered and the Co 3d band shifts to a lower energy. Since such Co 3d band largely hybridizes with the Ce 4f band, the Ce 4f band edge is lowered as a result of Y doping. Subsequently, the O 2p to Ce 4f band gap decreases with increasing Y concentration in the (Y, Co)-codoped CeO2.
Our results show that increasing the oxygen vacancy concentration by Y doping alone cannot modulate the band gap in the oxygen-vacancy-sensitive CeO2 nanocrystals. However, band gap narrowing via Y-induced oxygen vacancies can be achieved by incorporating a small amount of a triggering dopant element that generates an impurity band in the gap and largely hybridizes with the conduction band. In the present work, Co acts as a switch while Y plays the role of a modulator for tuning the band gap in the CeO2 host. The interplay of such switch and modulator codopants for band gap engineering can lead to many important applications, especially in environmental protection and solar energy harvesting.
Additional Information
How to cite this article: Wu, T. S. et al. Unconventional interplay between heterovalent dopant elements: Switch-and-modulator band-gap engineering in (Y, Co)-Codoped CeO2 nanocrystals. Sci. Rep. 5, 15415; doi: 10.1038/srep15415 (2015).
References
Corma, A., Atienzar, P., García, H. & Chane-Ching, J. Y. Hierarchically mesostructured doped CeO2 with potential for solar-cell use. Nat Mater. 3, 394–397 (2004).
Lawrence, N. et al. Defect Engineering in Cubic Cerium Oxide Nanostructures for Catalytic Oxidation. Nano Lett. 11, 2666–2671 (2011).
Liyanage, A. D., Perera, S. D., Tan, K., Chabal, Y. & Balkus, K. J. Jr. Synthesis, Characterization and Photocatalytic Activity of Y-Doped CeO2 Nanorods. ACS Catal. 4, 577–584 (2014).
Suzuki, T., Kosacki, I., Anderson, H. U. & Colomban, P. Electrical Conductivity and Lattice Defects in Nanocrystalline Cerium Oxide Thin Films. J. Am. Ceram. Soc. 84, 2007–2014 (2001).
Keating, P. R. L., Scanlon, D. O., Morgan, B. J., Galea, N. M. & Watson, G. W. Analysis of Intrinsic Defects in CeO2 Using a Koopmans-Like GGA+U Approach. J. Phys. Chem. C 116, 2443–2452 (2012).
Li, M. et al. Evidence of Oxygen Vacancies Enhancing the Room Temperature Ferromagnetism in CeO2−x Nanopowders. IEEE Trans. Magn. 44, 2708–2711 (2008).
Choudhury, B. & Choudhury, A. Lattice distortion and corresponding changes in optical properties of CeO2 nanoparticles on Nd doping. Curr. Appl. Phys. 13, 217–223 (2013).
Rey, J. F. Q. & Muccillo, E. N. S. Lattice parameters of yttria-doped ceria solid electrolytes. J. Eur. Ceram. Soc. 24, 1287–1290 (2004).
Langford, J. I. & Wilson, A. J. C. Scherrer after Sixty Years: A Survey and Some NewResults in the Determination of Crystallite Size. J. Appl. Cryst. 11, 102–113 (1978).
Wu, T. S. et al. Correlation Between Oxygen Vacancies and Magnetism in Mn-doped Y2O3 Nanocrystals Investigated by Defect Engineering Techniques. Appl. Phys. Lett. 101, 022408 (2012).
Soo, Y. L. et al. Thermal annealing and grain boundary effects on ferromagnetism in Y2O3:Co diluted magnetic oxide nanocrystals. Appl. Phys. Lett. 98, 031906 (2011).
Newville, M., Lïvins, P., Yacoby, Y., Rehr, J. J. & Stern, E. A. Near-edge x-ray-absorption fine structure of Pb: A comparison of theory and experiment. Phys. Rev. B 47, 14126–14131 (1993).
Rehr, J. J., Mustre de Leon, J., Zabinsky, S. I. & Albers, R. C. Theoretical x-ray absorption fine structure standards. J. Am. Chem. Soc. 113, 5135–5140 (1991).
Soo, Y. L. et al. X-ray excited luminescence and local structures in Tb-doped Y2O3 nanocrystals. J. Appl. Phys. 83, 5404–5409 (1998).
Kubelka, P. & Munk, F. Ein Beitrag zur Optik der Farbanstriche. Z. Tech. Phys. (Leipzig) 12, 593–601 (1931).
Tauc, J. & Menth, A. States in the gap. J. Non-Cryst. Solids 8, 569–585 (1972).
Zhou, X.-D. & Huebner W. Size-induced lattice relaxation in CeO2 nanoparticles. Appl. Phys. Lett. 79, 3512–3514 (2001).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P. et al. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Andersson, D. A., Simak, S. I., Johansson, B., Abrikosov, I. A. & Skorodumova, N. V. Modeling of CeO2, Ce2O3 and CeO2−x in the LDA+U formalism. Phys. Rev. B 75, 035109 (2007).
Loschen, C., Carrasco, J., Neyman, K. M. & Illas, F. First-principles LDA+U and GGA+U study of cerium oxides: Dependence on the effective U parameter. Phys. Rev. B 75, 035115 (2007).
Castleton, C. W., Kullgren, J. & Hermansson, K. Tuning LDA+U for electron localization and structure at oxygen vacancies in ceria. J. Chem. Phys. 127, 244704 (2007).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Author information
Authors and Affiliations
Contributions
T.S.W. and Y.L.S. conceived and designed the experiments. T.S.W. prepared the materials. T.S.W., H.D.L., Y.W.C., S.F.C., C.H.C., C.W.P., J.F.L., S.L.C. and Y.L.S. planned and performed the synchrotron radiation x-ray measurements. Y.S.S. and C.H.L. performed the energy gap measurements. T.S.W. and H.T.J. performed the DFT calculations. T.S.W. and Y.L.S. co-wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wu, T., Li, H., Chen, Y. et al. Unconventional interplay between heterovalent dopant elements: Switch-and-modulator band-gap engineering in (Y, Co)-Codoped CeO2 nanocrystals. Sci Rep 5, 15415 (2015). https://doi.org/10.1038/srep15415
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15415
This article is cited by
-
Formation of Co–O bonds and reversal of thermal annealing effects induced by X-ray irradiation in (Y, Co)-codoped CeO2 nanocrystals
Scientific Reports (2022)
-
Enhancement of catalytic activity by UV-light irradiation in CeO2 nanocrystals
Scientific Reports (2019)
-
Dramatic band gap reduction incurred by dopant coordination rearrangement in Co-doped nanocrystals of CeO2
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
