
Abstract
Host cells orchestrate the production of IFN-β upon detecting invading viral pathogens. Here, we report that Ring finger protein 166 (RNF166) potentiates RNA virus-triggered IFN-β production. Overexpression of RNF166 rather than its homologous proteins RNF114, RNF125 and RNF138, enhanced Sendai virus (SeV)-induced activation of the IFN-β promoter. Knockdown of endogenous RNF166, but not other RNFs, inhibited the IFN-β production induced by SeV and encephalomyocarditis virus. RNF166 interacted with TRAF3 and TRAF6. SeV-induced ubiquitination of TRAF3 and TRAF6 was suppressed when endogenous RNF166 rather than RNF114/138 was knocked down. These findings suggest that RNF166 positively regulates RNA virus-triggered IFN-β production by enhancing the ubiquitination of TRAF3 and TRAF6.
Similar content being viewed by others


Introduction
Innate immunity provides a robust first line of defense against invading pathogens. After detecting invading viruses, host cells initiate several signaling cascades to generate type I interferons (IFNs) such as IFN-β and IFN-α. Type I IFNs activate the JAK-STAT pathway, resulting in expression of hundreds of interferon-stimulated genes, which can target every stage of the viral life-cycle and protect host cells from invading viruses1.
Members of the RLR family, including retinoic acid inducible gene-I (RIG-I), melanoma differentiation-associated gene 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2), are located in the cytoplasm to monitor viral RNA2. Upon viral infection, the helicase domain of RIG-I and MDA5 sense viral RNA that bears a 5′-triphosphate group that is lacking in host mRNA3,4. After binding viral RNA, RIG-I and MDA5 undergo conformational changes as well as modifications with K63-linked polyubiquitin chains by TRIM25 and REUL (also known as Riplet or RNF135)5,6,7,8. Ubiquitinated RIG-I and MDA5 interact with VISA (also named MAVS, Cardif or IPS-1) and this results in aggregation of the latter9,10,11,12. VISA polymers then recruit TRAFs such as TRAF3 and TRAF6 to promote the ubiquitination reaction which is critical for recruiting IKK and TBK1 to the VISA signaling complex13. IKK and TBK1 phosphorylate VISA, resulting in binding of VISA to the conserved, positively-charged surfaces of IRF3, thereby recruiting IRF3 for phosphorylation and activation14.
The identity of the cytoplasmic DNA sensor remained unresolved until researchers recently identified cyclic GMP-AMP synthase (cGAS) as a new viral DNA sensor15,16,17. Upon DNA viral infection, cGAS directly binds to DNA and releases its catalytic pocket to ATP and GTP for the generation of 2′3′-cGAMP18,19,20,21,22. cGAMP binds to and activates STING to assemble a punctate structure that contains TBK1. TBK1 then phosphorylates STING and this is followed by the recruitment of IRF3 to STING for phosphorylation and activation14.
Ubiquitination plays a critical role in the RNA virus-induced innate immune response. As noted above, K63 ubiquitination of RIG-I triggered by TRIM25 and REUL is indispensable for its activation5,6,7,8, while Ring-finger protein 125 (RNF125) and c-Cbl catalyze the K48-linked ubiquitination of RIG-I and negatively regulate RIG-I-mediated antiviral activity23,24. Ubiquitin carboxyl-terminal hydrolase CYLD, a de-ubiquitination enzyme, physically interacts with RIG-I and removes its K63-linked polyubiquitin chains to attenuate antiviral activity25. VISA polymers can also recruit ubiquitin ligase family members, multiple TRAFs, through different TRAF-binding motifs to promote K63-linked ubiquitination, thereby recruiting NEMO to the VISA complex, which turns on TBK1 and IKK, resulting in the activation of IRF3 and NF-κB13. In addition, cIAP1/2 acts as a positive regulator by enhancing RNA virus-mediated K63-linked ubiquitination of TRAF3/6, while OTUB1/2 plays an opposite role via deubiquitinating TRAF3/626,27.
In this report, we show that Ring-finger protein 166 (RNF166) potentiates RNA virus-induced IFN-β production via enhancing the ubiquitination of TRAF3 and TRAF6. These findings broaden our understanding of the mechanisms by which RLR signaling is positively regulated upon viral infection.
Results
RNF166 rather than its homologous proteins potentiates RNA virus-induced IFN-β production
RNF166 is closely related to RNF125, which has been reported to negatively regulate RIG-I- mediated anti-RNA virus signaling by conjugating ubiquitin chains to RIG-I and leading to the degradation of RIG-I by the proteasome23. RNF125 and its homologous proteins RNF114, RNF138 and RNF166 form a subfamily of small C3HC4 RING ubiquitin ligases28, so we investigated whether RNF114/138/166 also play a role in RNA virus-induced IFN-β production. We transfected plasmids that encoded RNF114, RNF125, RNF138 and RNF166 into HEK293T cells to perform reporter assays. We found that overexpression of RNF166 but not it’s homologous RNF114, 125 and 138, potentiated Sendai virus (SeV)-induced activation of the IFN-β promoter. However, RNF166 had no apparent effect on the overexpression of cGAS and the STING-induced activation of the IFN-β promoter (Fig. 1A), suggesting that RNF166 specifically enhances RNA but not DNA virus-induced IFN-β production. Overexpression of RNF166 can also enhance the transcription of Interferon-stimulated genes (ISGs) like ISG15 and MX1 (Fig. 1B).

RNF166 rather than RNF114, RNF125, or RNF138 potentiates RNA virus-induced IFN-β production.
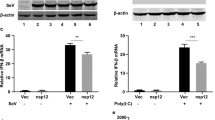
(A) RNF166 positively regulates SeV- but not cGAS- and STING-induced activation of the IFN-β promoter. 293T cells (2 × 105) were transfected with IFN-β reporter (50 ng), pRL-TK Renilla luciferase plasmid (50 ng) and the expression plasmids indicated above. 12 h after transfection, cells were infected with SeV or left uninfected and luciferase assays were performed 24 h after infection. (B) RNF166 enhances the transcription of ISG15 and MX1. 293T cells (2 × 106) were transfected with empty vector or expression plasmid of RNF166 (2 μg), 24 h after transfection, the mRNA of ISG15 and MX1 were detected by Q-PCR. (C) HEK293T cell pools with stable knockdown RNF166 were generated by shRNA. Knockdown efficiency was determined by RT-PCR (left) and Q-PCR (right). (D) SeV-mediated activation of the IFN-β reporter was inhibited when the endogenous RNF166 was stably knocked down. Transfection and luciferase assays were performed as in (A). (E) SeV-induced IFN-β production by 293T-shRNF166-#4 and #5 cells was lower than by 293T-shGFP cells. Cells (1 × 106) were infected with SeV. 12 h after infection, cells were analyzed by Q-PCR (left) and the supernatants were collected for IFN-β bioassays (right). (F) HEK293T cell pools with stable knockdown of RNF114, RNF125 and RNF166 were generated using shRNA. Knockdown efficiency was determined by RT-PCR (left) and Q-PCR (right). (G) SeV-induced IFN-β production by cell pools with stable knockdown of RNF114, RNF125 and RNF138. Cells were infected and analyzed as in (E). (H) The knockdown efficiency of RNF166 in HeLa cells was determined by RT-PCR. (I–L) SeV-, EMCV-infection, poly (I:C)-transfection(4 ug) and Influenza A-infection, rather than dsVACV-transfection(4 ug) induced production of IFN-β by HeLa-shRNF166-#4 and #5 cells were lower than by HeLa-shGFP cells. Q-PCR and bioassays were performed at 12 h after treatment as in (E). Each graph represents the mean ± SD of three independent experiments done in triplicate. ***indicates P < 0.001; **indicates P < 0.01; ns (not significant) indicates P > 0.05.
To determine whether endogenous RNF166 is involved in anti-RNA virus signaling, we generated stable RNF166-knockdown cell pools using shRNA plasmids that targeted five sites on human RNF166 mRNA. Two shRNA plasmids (#4 and #5) markedly inhibited the expression of endogenous RNF166 mRNA in HEK293T cells, whereas the #1, #2 and #3 shRNA plasmids had little effect on RNF166 mRNA (Fig. 1C). We found that knockdown of RNF166 significantly inhibited the activation of the IFN-β promoter, transcription of IFN-β mRNA and the secretion of IFN-β triggered by SeV infection (Fig. 1D,E). We also generated stable RNF114-, RNF125- and RNF138-knockdown HEK293T cell pools to determine their functions (Fig. 1F). Consistent with a previous report that RNF125 acts as a negative regulator of the RIG-I-mediated signaling pathway23, knockdown of RNF125 slightly enhanced the SeV-elicited IFN-β production compared to the control cell pool, while knockdown of RNF114 clearly enhanced the SeV-induced IFN-β production and knockdown of RNF138 had no appreciable effect (Fig. 1G).
We further generated stable RNF166-knockdown cell pools with HeLa cells and obtained similar results; the production of IFN-β induced by SeV, encephalomyocarditis virus (EMCV) and human influenza A virus infection, as well as poly (I:C)-transfection notably decreased when endogenous RNF166 expression was knocked down (Fig. 1H–K). However, knockdown of RNF166 had no effect on DNA analog-induced activation of IFN-β (Fig. 1L). These results suggested that RNF166 rather than its homologous proteins physiologically potentiates RNA virus-induced IFN-β production.
RNF166 targets TRAF3 and TRAF6 to potentiate VISA-mediated antiviral signaling
We next determined which molecules are targets of RNF166 in the anti-RNA virus signaling pathway. In reporter assays, overexpression of RNF166 potentiated VISA-, but not the downstream kinase TBK1-mediated activation of the IFN-β promoter (Fig. 2A). Repoter Assay and Bioassay results showed that RNF166 but not the other RNFs potentiated VISA-mediated transcription and secretion of IFN-β, while the overexpression of RNF114 or RNF125 had inhibitory effects (Fig. 2B). Accordingly, knockdown of RNF166 inhibited VISA- but not TBK1-mediated activation of the IFN-β promoter (Fig. 2C). These data suggested that RNF166 acts on signaling components that are downstream of VISA and upstream of TBK1.

RNF166 targets TRAF3 and TRAF6 to potentiate VISA-mediated antiviral signaling.
(A) RNF166 potentiates VISA- but not TBK1-mediated activation of the IFN-β promoter. 293T cells (2 × 105) were transfected with IFN-β reporter (50 ng), pRL-TK Renilla luciferase plasmid (50 ng) and expression plasmids for VISA or TBK1 together with an empty vector and RNF166 construct (50, 100 and 200 ng). Luciferase assays were performed 24 h after transfection. (B) RNF166 rather than RNF114, RNF125 and RNF138 potentiates the VISA-mediated activation of the IFN-β promoter and production of IFN-β. 293T cells (2 × 105) were transfected with the indicated plasmids. 24 h after transfection, the supernatants collected for IFN-β bioassays (right); Luciferase assays (left) were performed as in (A). (C) VISA- rather than TBK1-mediated activation of the IFN-β promoter was attenuated in 293T-shRNF166-#4 and #5 cells compared with 293T-shGFP cells. Luciferase assays were performed as in (A). (D) Overexpressed RNF166 interacts with overexpressed VISA, TRAF3 and TRAF6. 293T cells (1 × 107) were transfected with the indicated plasmids (5 μg each). Cell lysates were immunoprecipitated (IP) with anti-HA (αHA). 24 h after transfection the immunoprecipitates and whole-cell lysates (WCL) were analyzed by western blot (IB) with anti-HA and anti-Flag antibody. (E) RNF166 co-localized with TRAF6 and TRAF3 but not VISA in HeLa cells. HeLa cells were transfected indicated expression plasmids (1 μg each). Immunofluorescent staining was performed with anti-Flag (red), anti-HA (green) and DAPI (blue). (F) Effects of SeV infection on the interaction between overexpressed RNF166 and endogenous TRAF3 and TRAF6. 293T cells (5 × 107) were transfected with Flag-RNF166 plasmids (5 μg each). 18 h after transfection, cells were infected with SeV for the indicated times. The cell lysates were immunoprecipitated (IP) and analyzed by western blot (IB) with the indicated antibodies. (G) RNF166 did not potentiate the VISA-mediated activation of the IFN-β promoter in 293T-shTRAF3 and 293T-shTRAF6 cells. Luciferase assays were performed as in (A). Each graph represents the mean ± SD of three independent experiments done in triplicate. ***indicates P < 0.001; **indicates P < 0.01; ns (not significant) indicates P > 0.05.
We next used co-immunoprecipitation to determine whether RNF166 interacts with VISA and its downstream components. Overexpressed RNF166 markedly associated with TRAF3 and TRAF6 in 293T cells and also interacted with VISA, but had no detectable interactions with RIG-I, MDA5, TBK1 and IRF3 (Fig. 2D). RNF166 was subsequently shown to co-localize with TRAF3 and TRAF6 in co-transfected HeLa cells (Fig. 2E). Overexpressed RNF166 localized predominantly in the cytosol as dots which were probably aggregates of RNF166 protein. Double immunofluorescent staining showed that overexpressed TRAF6/3, especially TRAF6, had a similar distribution pattern and overlapped with RNF166, while we did not detect co-localization between RNF166 and VISA.
To define these interactions under physiological conditions, we set out to determine associations between endogenous RNF166 and the targets. As we could not obtain an effective antibody to detect endogenous RNF166 through preparation or commercial purchase, we performed immunoprecipitation between overexpressed RNF166 and endogenous TRAF3 and TRAF6 with or without SeV infection. Overexpressed RNF166 weakly interacted with endogenous TRAF3 and TRAF6 and SeV infection greatly enhanced these interactions (Fig. 2F). These data suggested that RNF166 associates with TRAF3 and TRAF6 during viral infection.
To further clarify whether RNF166 targets TRAF3 and TRAF6, we used shRNA to knockdown endogenous TRAF3 or TRAF6 and found that RNF166 no longer potentiated VISA-induced IFN-β activation when TRAF3 or TRAF6 expression was suppressed (Fig. 2G). These data further supported the idea that RNF166 targets TRAF3 and TRAF6 to potentiate RNA virus-induced IFN-β production.
Both TRAF3 and TRAF6 play a critical role in SeV-induced interferon-β production in HEK293T cells
That TRAF3 acts as a critical adaptor in RIG-I-mediated antiviral signaling has been demonstrated by several studies29,30, but its role has been doubted recently13. So we then determined whether TRAF3 or TRAF6 plays a critical role in SeV-induced IFN-β production by using shRNA to stably knock down endogenous TRAF3 or TRAF6 in HEK293T cells (Fig. 3A). We found that VISA-mediated activation of the IFN-β promoter, SeV-induced transcription and the secretion of IFN-β were markedly reduced (Figs 2G and 3B) and SeV-induced phosphorylation and dimerization of IRF3 was apparently inhibited when TRAF3 or TRAF6 expression was suppressed (Fig. 3C).

Both TRAF3 and TRAF6 play a critical role in SeV-induced signaling in HEK293T cells.
(A) 293T cell pools with stable knockdown of TRAF3, TRAF6 and VISA were generated by shRNA. Knockdown efficiency was analyzed by western blot with anti-TRAF3, TRAF6 and VISA antibodies. (B) SeV-induced IFN-β production was reduced when endogenous TRAF3, TRAF6, or VISA was stably knocked down. Indicated cells (5 × 105) were infected with SeV and IFN-β levels were estimated by Q-PCR (B, left) and bioassay (B, right) at the indicated times. (C) IRF3 dimer and p-IRF3 were attenuated in 293T-shTRAF3, 293T-shTRAF6 and 293T-shVISA cells infected with SeV. Indicated cells (5 × 105) were infected with SeV for the indicated times and lysates were analyzed by western blot with anti-p-IRF3 and anti-GADPH or by native western blot with anti-IRF3. (D) TRAF3 and TRAF6 knockout HEK293T cell lines were generated by CRISPER/CAS9. Knockout cells were analyzed by western blot with anti-TRAF3, TRAF6 and VISA antibodies. (E) p-IRF3 was attenuated in TRAF3-knockout (KO) 293T, TRAF6-KO 293T and VISA-KO 293T cells infected with SeV. Indicated knockout cells (5 × 105) were infected with SeV and lysates were analyzed by western blot with anti-p-IRF3 antibody. (F) SeV-induced IFN-β was reduced when endogenous TRAF3, TRAF6 and VISA were knocked out. Q-RT-PCR (left) and IFN-β bioassay (right) were performed as in (B). Each graph represents the mean ± SD of three independent experiments done in triplicate. ***indicates P < 0.001; **indicates P < 0.01; ns (not significant) indicates P > 0.05.
To further determine the function of TRAF3 and TRAF6, we generated TRAF3 or TRAF6 knockout HEK293T cell lines using the CRISPR/Cas9 system (Fig. 3D). Phosphorylation of IRF3 triggered by SeV-infection was greatly inhibited and the production of IFN-β was blocked when TRAF3 or TRAF6 was deleted (Fig. 3E,F). These results suggested that both TRAF3 and TRAF6 play a critical role in SeV-induced IFN-β production in HEK293T cells.
Functional domain mapping of RNF166
RNF166 contains a RING domain, a zinc finger domain and an ubiquitin-binding domain (UIM) (Fig. 4A). To dissect the functional role of these domains, we generated three deletion mutants and assessed their ability to up-regulate SeV-induced IFN-β production. We found that the presence of an intact RING domain is essential for RNF166 function, as overexpression of a RING deletion mutant (RNF166 ΔRING) failed to enhance, but rather inhibited the activation of the IFN-β promoter induced by SeV and the overexpression of VISA, while the UIM domain deletion mutant (RNF166 ∆UIM) had no apparent effect (Fig. 4B,C). Consistently, on infection with NDV-eGFP (Newcastle disease virus-enhanced green fluorescent protein), we found that overexpression of RNF166 in HEK293T cells rendered them remarkably resistant to NDV infection and reduced the levels of NDV-eGFP-positive cells, while the RING deletion mutant had the opposite effect (Fig. 4D); and the same results were obtained when RNF166 or RNF166 ΔRING were co-expressed with VISA. Also we found overexpression of RNF166 can inhibit the replication of SeV (measured by mRNA level of SeV P protein), while the RING delete mutant enhanced the proliferation of SeV (Fig. 4E). These data indicated that the RING domain is indispensable for the ability of RNF166 to up-regulate cellular anti-RNA virus activity.

Functional domain mapping of RNF166.
(A) Schematic structures of RNF166 and the deletion mutants used in this work. (B,C) Effects of truncations of RNF166 on SeV-induced (B) and VISA-mediated (C) activation of the IFN-β promoter. Transfection and luciferase assays were performed as above. (D) Effects of RNF166 and the RNF166 RING deletion mutant on VISA-mediated antiviral activity. 293T cells (5 × 105) were transfected with the indicated plasmids (1 μg). At 20 h after transfection, cells were infected with NDV-eGFP at an MOI of 0.0001 and at 40 h after infection, viral replication was determined by fluorescence microscopy. The GFP-positive cells were counted and the percentage was calculated. (E) Effects of RNF166 and the RNF166 RING deletion mutant on replication of SeV. 293T cells (5 × 105) were transfected with the indicated plasmids (1 μg). At 12 h after transfection, cells were infected with SeV and at 24 h after infection, mRNA of P protein was determined by Q-PCR. (F) RNF166 interacted with TRAF3 and TRAF6 via its zinc-finger domain. Transfection and immunoprecipitation (IP) were performed as in Fig. 2 (D). Each graph represents the mean ± SD of three independent experiments done in triplicate. ***indicates P < 0.001; **indicates P < 0.01; ns (not significant) indicates P > 0.05.
We further performed co-immunoprecipitation to detect which domain of RNF166 is required for interactions with TRAF3 and TRAF6. The results showed that the RING deletion and UIM deletion mutants interacted with TRAF3/6 just like full-length RNF166, while the mutant carrying the RING domain only did not (Fig. 4F), suggesting RNF166 interacts with TRAF3 and TRAF6 via its zinc finger domain and the RING domain is necessary for its positive regulatory function.
RNF166 enhances RNA virus-induced ubiquitination of TRAF3 and TRAF6
It is known that the RING domain is the critical functional domain for E3 ubiquitin ligase and polyubiquitination of TRAF3 and TRAF6 are important for RLR signaling13,27, so we suggested that RNF166 could affect the ubiquitination of TRAF3 and TRAF6. We found overexpressed RNF166 enhanced the ubiquitination of TRAF3 and TRAF6 in co-transfection experiments, consistent with previous results and RNF166 lost this effect when the RING domain was deleted (Fig. 5A).

RNF166 enhances virus-mediated ubiquitination of TRAF3/TRAF6.
(A) Overexpression of RNF166 enhanced the ubiquitination of overexpressed TRAF3/6. 293T cells (1 × 107) were transfected with the indicated plasmids (5 μg each). Cell lysates were immunoprecipitated (IP) with anti-Flag (αF). The immunoprecipitates and whole-cell lysates (WCL) were analyzed by western blot (IB) with anti-HA and anti-Flag antibodies. (B,C) Knockdown of RNF166 inhibited SeV-mediated WT- and K63- but not K48-linked ubiquitination of endogenous TRAF3 (B) and TRAF6 (C). Indicated cells (3 × 107) were transfected with WT ubiquitin, K63O ubiquitin, or K48O ubiquitin (5 μg each). At 18 h after transfection, cells were infected with SeV for the indicated times. The lysates were immunoprecipitated with anti-TRAF3 (αTRAF3) or anti-TRAF6 (αTRAF6) antibody. The immunoprecipitates and whole-cell lysates (WCL) were analyzed by western blot (IB) with the indicated antibody. (D,E) Knockdown of RNF114 (D) and RNF138 (E) did not inhibit the SeV-mediated ubiquitination of endogenous TRAF3 and TRAF6. Transfection and immunoprecipitation were performed as in (B).
We then found that the SeV-induced ubiquitination of endogenous TRAF3 and TRAF6 was notably inhibited when RNF166 expression was suppressed (Fig. 5B,C). We further found that K63-linked rather than K48-linked ubiquitination of TRAF3 and TRAF6 was decreased upon SeV infection when RNF166 was knocked down (Fig. 5B,C). However, we did not find an apparent effect of knockdown of endogenous RNF114 and RNF138 on SeV-induced ubiquitination of TRAF3 and TRAF6 (Fig. 5D,E). These data suggested that endogenous RNF166 rather than its homologous proteins RNF114 and RNF138 specifically enhances the SeV-induced ubiquitination of TRAF3 and TRAF6.
Discussion
Virus-triggered production of IFN-β is critical for the antiviral immune response and is delicately regulated in space and time by various molecules and distinct mechanisms. Ubiquitination has emerged as a critical role in forming signaling complexes and transducing signals downstream13.
RNF125 has been reported to negatively regulate RIG-I-mediated antiviral activity via conjugating ubiquitin chains to RIG-I and MDA5, leading to their degradation by the proteasome. However, the functions of its homologous proteins RNF114, RNF125 and RNF138 in innate immune signaling pathways remain elusive. Here, we report that RNF166 rather than RNF114, RNF138 and RNF125 potentiates the RNA virus-induced production of IFN-β. Several lines of evidence support this finding. First, overexpressed RNF166 potentiated the SeV- and VISA-mediated induction of IFN-β. Second, knockdown of endogenous RNF166 by shRNA reduced the RNA virus- and dsRNA analog-induced production of IFN-β. Third, RNF166 interacted with endogenous TRAF3 and TRAF6 and these interactions were enhanced upon viral infection. Furthermore, we found that the potentiation of the antiviral effect of RNF166 was mediated by enhancing the virus-induced ubiquitination of TRAF3 and TRAF6. Notably, endogenous RNF114 and RNF138 had no apparent effect on the virus-induced ubiquitination of TRAF3 and TRAF6 and this finding may explain why RNF114 and RNF138 do not enhance SeV-induced antiviral activity while RNF166 did.
TRAF3 and TRAF6 play a vital role in the RIG-I-mediated signaling pathway29,30,31. Therefore, both molecules are functionally regulated by cellular components to control the immune response and viral proteins to evade the host immune system. These regulatory mechanisms include interfering with the interaction between VISA and TRAF3/632,33,34, deubiquitinating TRAF3/626,32,33,34,35 and disrupting the binding of downstream molecules to TRAF3/636,37,38,39. Although a recent study has shown that TRAF3 is dispensable for the RNA virus-induced activation of TBK1 and IRF313, our data suggest that TRAF3 is critical for the SeV-elicited production of IFN-β in HEK293T cells. We cannot exclude the possibility that the differences in the cell lines and experimental methods we used explains the different conclusions. However, we agree that TRAF6 is a critical component downstream of VISA13.
In light of our study, RNF166 plays an important role in RNA virus-triggered IFN-β production by enhancing the ubiquitination of TRAF3 and TRAF6. We noted that expression of a mutant with the RING domain deleted, RNF166-∆RING, markedly suppressed VISA-mediated activation of the IFN-β promoter and anti-NDV activity. These data indicate that, as a dominant-negative mutant, RNF166∆RING competes with endogenous RNF166 to bind with TRAF3 or TRAF6, so they cannot be efficiently ubiquitinated and blocks VISA signaling. However, STING has no apparent TRAF-binding motifs that are critical for recruiting TRAFs and can directly recruit IRF3 and TBK1 for activation via its carboxyl terminal region40; this may explain why RNF166 does not potentiate the cGAS- and STING-induced activation of the IFN-β promoter.
RNF114 was first identified as a psoriasis-susceptibility gene41,42. Later study revealed that overexpressed RNF114 enhances NF-ΚB and IRF3 reporter activity and increases type I IFN mRNA levels43. However, the analysis of cells with RNF114 knockdown yielded a heterogeneous set of results that may have been due to the presence of different populations within the polyclonal cell lines or a redundant role of RNF11443. Recently, another independent study revealed that RNF114 acts as a negative regulator of NF-κB-dependent transcription by stabilizing the A20 protein and IκBα44. Our data from overexpression and knockdown experiments indicated that RNF114 acts as a negative regulator of SeV-induced IFN-β signaling. A20 has been demonstrated to negatively regulate RIG-I-mediated signaling in several independent studies45,46,47, so it is possible that RNF114 depends on A20 to play negative regulatory roles. However, further investigations are needed to confirm the function of RNF114 in the innate immunity signaling pathway.
Although RNF114, RNF125, RNF138 and RNF166 have similar domain structures, their functions in SeV-induced signaling are quite diverse, including both positive and negative effects. Future investigations into their structures are expected to elucidate the molecular mechanisms underlying their different functions in the innate immunity signaling pathway.
Methods
Reagents and cell lines
Mouse antibodies against Flag and HA epitopes (Sigma-Aldrich), rabbit IgG (Sigma-Aldrich), IRDye800-conjugated anti-mouse and anti-rabbit IgG (Rockland Immunochemicals), rabbit polyclonal antibody against TRAF3 (Proteintech), rabbit polyclonal antibody against TRAF6 (Santa Cruz Biotechnology), mouse monoclonal antibody against GADPH (Abmart), poly(I:C) (Pharmacia), mouse polyclonal antibody against VISA (Hong-Bing Shu, Wuhan University, China), NDV-enhanced eGFP (Cheng Wang, Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China), Human Influenza A virus (Xin Ye, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China), EMCV (Xin Guo, China Agriculture University, Beijing, China) and HT1080 stably-transfected with ISRE luciferase construct (2fTGH) (Zheng-Fan Jiang, Peking University, China) were from the indicated sources. HEK293T, HeLa and 2fTGH cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Life Technologies) and 100-units/ml penicillin/streptomycin.
Constructs
Mammalian expression plasmids for Flag- or HA-tagged RNF114, RNF125, RNF138, RNF166 and RNF166 truncations were constructed using standard molecular biology techniques. Mammalian expression plasmids for Flag-RIG-I, -MDA5, -VISA, -TRAF3, -TRAF6, -TBK1, -IRF3, HA-ubiquitin, -K48O ubiquitin, -K63O ubiquitin (Lys-48- and Lys-63-only ubiquitin mutants; all lysine residues except Lys-48 and Lys-63 were mutated to arginine) and IFN-β promoter luciferase reporter plasmids were provided by Dr. Hong-Bing Shu (Wuhan University, Wuhan, China).
Transfection and Luciferase Assay
HEK293T cells were seeded onto 24-well dishes and transfected the next day with polyethylenimine (Electron Microscopy Sciences). To normalize for transfection efficiency, 50 ng of pRL-Tk Renilla luciferase reporter plasmid was added to each transfection. About 18 h after transfection, assays were performed using a dual-specific luciferase assay kit (Promega). Firefly luciferase activity was normalized based on Renilla luciferase activity. All reporter assays were repeated at least three times.
Co-immunoprecipitation and western blot analysis
For transient transfection and immunoprecipitation experiments, HEK293T cells (2 × 106) were transfected with the indicated plasmids for 20 h. The transfected cells were lysed in 0.5 ml lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10 mg/ml aprotinin, 10 mg/ml leupeptin and 1 mM phenylmethylsulfonyl fluoride). For each immunoprecipitation, a 0.4-ml aliquot of lysate was incubated with 0.5 μg of the indicated antibody and 25 μl of a 1:1 slurry of protein A-Sepharose (GE Healthcare) for 4 h. The Sepharose beads were washed three times with 1 ml lysis buffer. The precipitates were analyzed by western blotting with the indicated antibodies and visualized by incubation with IRDye800-conjugated secondary antibodies using an Odyssey infrared imaging system (Licor Inc.).
Immunofluorescent staining
Cells were fixed in ice-cold methanol for 20 min at −20 °C, rehydrated three times with phosphate-buffered saline (PBS) and blocked in 5% bovine serum albumin–PBS for 30 min. The cells were stained with primary antibody in blocking buffer for 1 h at 37 °C, rinsed in PBS and stained again with Alexa Fluor 488-labeled rabbit anti-mouse IgG or Alexa Fluor 594-labeled goat anti-rabbit IgG (Life Technologies) for 1 h at 37 °C. The cells were then rinsed with PBS containing 4′, 6-diamidino-2-phenylindole (DAPI) and mounted. The cells were observed under a confocal microscope (LSM 710 NLO and DuoScan System, Zeiss).
Q-PCR
mRNA was isolated from cells using TRIzol reagent (Tiangen Biotech) and reverse-transcribed into cDNA with reverse transcriptase (Fermentas). cDNA was amplified on an ABI7300 Detection System (Applied Biosystems) using SYBR Green PCR Master Mix (Takara) according to the manufacturer’s instructions and data were normalized by the level of β-actin in each individual sample. The 2−ΔΔCt method was used to calculate relative expression changes. The gene-specific primers for Q-PCR were: IFN-β: 5′-AGGACAGGATGAACTTTGAC-3′ and 5′-TGATAGACATTAGCCAGGAG-3′; β-actin: 5′-ACGTGGACATCCGCAAAGAC-3′ and 5′-CAAGAAAGGGTGTAACGCAACTA-3′; RNF114: 5′-CGAGAGCACAGAGACTTC-3′ and 5′-AGGACAGTAAGGACAAGGA-3′; RNF125: 5′-CGTTCCTGTATTGCTACCA-3′ and 5′-CTTCTGACAAGTCCGAATATG-3′; RNF138: 5′-GGAACAGCAATAGGAGTGA-3′ and 5′-CTGGTAATCTGGCTAGGATC-3′; RNF166: 5′-AAGGTGACCCTGGCAAAGAT-3′ and 5′-GGGATAGGCTGTGATGTGGG-3′; ISG15: 5′-AGGCAGCGAACTCATCTTTG-3′ and 5′-CCAGCATCTTCACCGTCAG-3′; MX1: 5′-GTTTACCAGACTCCGACACGA-3′ and 5′- TTCCAGTGCCTTGATTTGCT -3′; P protein of SeV: 5′-TGTTATCGGATTCCTCGACGCAGTC-3′ and 5′-TACTCTCCTCACCTGATCGATTATC-3′.
IFN-β Bio-assay
The supernatants collected from HEK293T and HeLa cells were added to 2fTGH cells (stably transfected with the ISRE luciferase construct). After 5 h incubation, 2fTGH cells were lysed and firefly luciferase activity was measured using a luciferase assay kit (Promega).
Generation of stable knockdown cell pools
Plasmids encoding lentiviruses expressing short hairpin RNAs (shRNAs) were obtained from the library of the RNAi Consortium (Sigma-Aldrich). shRNF166-#1 (TRCN0000007800), #2 (TRCN0000007801), #3 (TRCN0000007802), #4 (TRCN0000007803), #5 (TRCN0000007804); shRNF114 (TRCN0000004485); shRNF125 (TRCN0000004234); shRNF138-#1 (TRCN0000033835), shRNF138-#2 (TRCN0000033837); shTRAF3 (TRCN0000034222); shTRAF6 (TRCN0000007352); and shVISA (TRCN0000148945) plasmids were purified and then transfected into HEK293T cells with a three-plasmid system to produce lentivirus. HEK293T and HeLa cells were plated onto 6-well plates at ~5 × 105 cells per well with 2 ml complete medium. One milliliter of the indicated lentivirus was added with 8 μg/ml polybrene. The plates were incubated for 48 h and the cells were selected with 2 μg/ml puromycin. Cells were collected for analysis 72 h after selection.
Generation of TRAF3/6 knockout cell lines
TRAF3 or TRAF6 knockout in HEK293T cells was generated by the CRISPR/CAS9 system containing pCDNA3.1-Cas9 and gRNA (a gift from Dr. Jian-Zhong Xi, Peking University, China) which has been described48. Five pairs of gRNA to target TRAF3 or TRAF6 were designed and transfected with Cas9 into HEK293T cells. Twenty-four hours after transfection, the genomic DNAs were extracted for PCR amplification and the indels confirmed by sequencing. The confirmed cell pools were diluted to generate monoclones which were tested by western blot using TRAF3 or TRAF6 antibody to identify gene-knockout cell lines. TRAF3-Cas9 target site: CCAGTTTTTGTCCCTGAACA; TRAF6-Cas9 target site: GTCTGAAAGTGACTGCTGTG. Sequencing primers of TRAF3: 5′-GGATTCTTGTGTTGCCTAA-3′ and 5′-TGGACTTCTTTAGGGCTTT-3′; sequencing primers of TRAF6: 5′-ATTACAACTGAGGTGCTTCT-3′and 5′-GCATCTGGTTCTGTTATAGGA-3′.
Statistical analysis
Two-way ANOVA analysis were used to analyze data. Experiments were repeated at least three times. Results were considered significant at p < 0.05.
Additional Information
How to cite this article: Chen, H.-W. et al. Ring finger protein 166 potentiates RNA virus-induced interferon-β production via enhancing the ubiquitination of TRAF3 and TRAF6. Sci. Rep. 5, 14770; doi: 10.1038/srep14770 (2015).
References
Schneider, W. M., Chevillotte, M. D. & Rice, C. M. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 32, 513–545 (2014).
Wu, J. & Chen, Z. J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 32, 461–488 (2014).
Andreas, P. et al. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science. 314, 997–1001 (2006).
Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 314, 994–997 (2006).
Gack, M. U. et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 446, 916–920 (2007).
Gao, D. et al. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One. 4, e5760, 10.1371/journal.pone.0005760 (2009).
Oshiumi, H., Matsumoto, M., Hatakeyama, S. & Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 284, 807–817 (2009).
Oshiumi, H. et al. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. 8, 496–509 (2010).
Kawai, T. et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 6, 981–988 (2005).
Meylan, E. et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 437, 1167–1172 (2005).
Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 122, 669–682 (2005).
Xu, L. G. et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 19, 727–740 (2005).
Liu, S. et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLIFE. 2, e00785, 10.7554/eLife.00785 (2013).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING and TRIF induces IRF3 activation. Science. 347, aaa2630–aaa2630 (2015).
Li, X. D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 341, 1390–1394 (2013).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 339, 826–830 (2013).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 498, 332–337 (2013).
Gao, P. et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 153, 1094–1107 (2013).
Kranzusch, P. J., Lee, A. S., Berger, J. M. & Doudna, J. A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368 (2013).
Li, X. et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 39, 1019–1031 (2013).
Zhang, X. et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 (2014).
Arimoto, K. et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. 104, 7500–7505 (2007).
Chen, W. et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell. 152, 467–478 (2013).
Friedman, C. S. et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936 (2008).
Li, S. et al. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem. 285, 4291–4297 (2010).
Mao, A. P. et al. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J Biol Chem. 285, 9470–9476 (2010).
Giannini, A. L., Gao, Y. & Bijlmakers, M. J. T-cell regulator RNF125/TRAC-1 belongs to a novel family of ubiquitin ligases with zinc fingers and a ubiquitin-binding domain. Biochem J. 410, 101–111 (2008).
Oganesyan, G. et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 439, 208–211 (2006).
Saha, S. K. et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 25, 3257–3263 (2006).
Hacker, H. et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 439, 204–207 (2006).
Wang, D. et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol. 85, 3758–3766 (2011).
Wang, S., Wang, K., Li, J. & Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J Virol. 87, 11851–11860 (2013).
Zhong, H. et al. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS One. 8, e80976, 10.1371/journal.pone.0080976 (2013).
Peng, Y., Xu, R. & Zheng, X. HSCARG negatively regulates the cellular antiviral RIG-I like receptor signaling pathway by inhibiting TRAF3 ubiquitination via recruiting OTUB1. PLoS Pathog. 10, e1004041, 10.1371/journal.ppat.1004041 (2014).
Alff, P. J., Sen, N., Gorbunova, E., Gavrilovskaya, I. N. & Mackow, E. R. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J Virol. 82, 9115–9122 (2008).
Charoenthongtrakul, S., Gao, L., Parvatiyar, K., Lee, D. & Harhaj, E. W. RING finger protein 11 targets TBK1/IKKi kinases to inhibit antiviral signaling. PLoS One. 8, e53717, 10.1371/journal.pone.0053717 (2013).
Ng, M. H. et al. MIP-T3 is a negative regulator of innate type I IFN response. J Immunol. 187, 6473–6482 (2011).
Siu, K. L. et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 284, 16202–16209 (2009).
Tanaka, Y. & Chen, Z. J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 5, ra20 (2012).
Capon, F. et al. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum Mol Genet. 17, 1938–1945 (2008).
Stuart, P. E. et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat Genet. 42, 1000–1004 (2010).
Bijlmakers, M. J., Kanneganti, S. K., Barker, J. N., Trembath, R. C. & Capon, F. Functional analysis of the RNF114 psoriasis susceptibility gene implicates innate immune responses to double-stranded RNA in disease pathogenesis. Hum Mol Genet. 20, 3129–3137 (2011).
Rodriguez, M. S. et al. The RING ubiquitin E3 RNF114 interacts with A20 and modulates NF-kappaB activity and T-cell activation. Cell Death Dis. 5, e1399 (2014).
Lin, R. et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 281, 2095–2103 (2006).
Maelfait, J. et al. A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog. 8, e1002570, 10.1371/journal.ppat.1002570 (2012).
Saitoh, T. et al. A20 Is a Negative Regulator of IFN Regulatory Factor 3 Signaling. J Immunol. 174, 1507–1512 (2005).
Chang, N. et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 23, 465–472 (2013).
Acknowledgements
The authors thank the Core Facilities at the School of Life Sciences, Peking University, for assistance with confocal microscopy and we are grateful to Dr. Xue-Mei Hao and Xiao-Cheng Li for help with capturing images. This work was supported by the National Natural Science Foundation of China (31170822 and 31470841).
Author information
Authors and Affiliations
Contributions
H.W.C. and D.Y.C. designed the experiments. H.W.C., Y.K.Y., H.X. and W.W.Y. implemented the experiments. H.W.C. and D.Y.C. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, HW., Yang, YK., Xu, H. et al. Ring finger protein 166 potentiates RNA virus-induced interferon-β production via enhancing the ubiquitination of TRAF3 and TRAF6. Sci Rep 5, 14770 (2015). https://doi.org/10.1038/srep14770
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14770
This article is cited by
-
New insights into the genetic predisposition of brucellosis and its effect on the gut and vaginal microbiota in goats
Scientific Reports (2023)
-
Multiple E3 ligases control tankyrase stability and function
Nature Communications (2023)
-
Early innate immune response triggered by the human respiratory syncytial virus and its regulation by ubiquitination/deubiquitination processes
Journal of Biomedical Science (2022)
-
The RING finger protein family in health and disease
Signal Transduction and Targeted Therapy (2022)
-
Regulation of antiviral innate immune signaling and viral evasion following viral genome sensing
Experimental & Molecular Medicine (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
