
Abstract
Jasmonates (JAs) are plant-specific key signaling molecules that respond to various stimuli and are involved in the synthesis of secondary metabolites. However, little is known about the JA signal pathway, especially in economically significant medicinal plants. To determine the functions of novel genes that participate in the JA-mediated accumulation of secondary metabolites, we examined the metabolomic and transcriptomic signatures from Salvia miltiorrhiza. For the metabolome, 35 representative metabolites showing significant changes in rates of accumulation were extracted and identified. We also screened out 2131 differentially expressed unigenes, of which 30 were involeved in the phenolic secondary metabolic pathway, while 25 were in the JA biosynthesis and signal pathways. Among several MeJA-induced novel genes, SmJAZ8 was selected for detailed functional analysis. Transgenic plants over-expressing SmJAZ8 exhibited a JA-insensitive phenotype, suggesting that the gene is a transcriptional regulator in the JA signal pathway of S. miltiorrhiza. Furthermore, this transgenic tool revealed that JAZ genes have novel function in the constitutive accumulation of secondary metabolites. Based on these findings, we propose that the combined strategy of transcriptomic and metabolomic analyses is valuable for efficient discovery of novel genes in plants.
Similar content being viewed by others

Introduction
Salvia miltiorrhiza is a well-known traditional Chinese medicine and considered as a model medicinal plant1,2. In Asia, its dry roots have been widely used for hundreds of years in the prevention and treatment of menstrual disorders, cardiovascular diseases, acute ischemic stroke diseases and inflammation3. The active ingredients of S. miltiorrhiza are divided into two main groups: lipid-soluble tanshinones and water-soluble phenolic acids. Both are secondary metabolites, levels of which are not only controlled by plant genotype but also enhanced by various biotic and abiotic stresses. The abiotic elicitor methyl jasmonate (MeJA) dramatically increases the biosynthesis of active ingredients in those species4,5,6. Therefore, this phenomenon might be exploited as a potential strategy for improving the production of valuable secondary metabolites by regulating the biosynthesis of jasmonates7. To data, numerous metabolites have been isolated and identified from S. miltiorrhiza8. In view of the economic value of its active ingredients, studies on the tanshinone9 and phenolic acid biosynthetic pathways10 are drawing considerable attention in recent years. Although several genes involved in metabolite biosynthesis are known to be induced by JA treatment, only a few have been identified through classical genetic screening.
Jasmonic acid (JA) and its MeJA derivative, collectively called jasmonates, are signaling molecules with regulatory functions in wound responses and some developmental processes11,12. The well-established JA biosynthesis pathway is shown in 13. Signal transduction begins with the recognition of JA by the Skp1/Cullin/F-box (SCF) complex through the ubiquitin-proteasome system. The Jasmonate Zim Domain (JAZ) proteins are targets of the SCFCOI1 complex and are subsequently subjected to proteasomal degradation in order to release positively acting transcription factors (TFs), such as MYC2 and MYB14,15,16. Exogenous application of MeJA results in strong activation of a large set of target genes and biochemical substances. Of interest to us are those involved in the JA-mediated biosynthesis of secondary compounds. We have obtained the proof of concept for this JA-dependent induction from studies of constitutive accumulation of caffeoylputrescine in tomato (Solanum lycopersicum)17, nicotine biosynthesis in tobacco (Nicotiana tabacum)18 and JA-induced anthocyanin formation19. In S. miltiorrhiza, Luo et al.20 reported 7 candidate cytochrome P450s which may be involved in the tanshinone and phenolic acid biosynthesis by investigating MeJA-induced gene expression profiles. However, JA-induced signal pathway and its connection with the downstream biosynthesis of active ingredients are still need to further investigate.
Integrated analyses of the metabolomes and transcriptomes of Arabidopsis21 and tomato22 have already led to the discovery of several new genes with special functions. This approach was also utilized to characterize the inducible nature of tanshinone production in hairy root cultures of S. miltiorrhiza23. These reports strengthened our confidence that such approaches maybe effective in comprehensive analyses of JA-induced gene expression and metabolite accumulations in S. miltiorrhiza. Furthermore, after previously combining the EST database with Solexa sequencing, we have obtained 57,754 unigenes, which comprises the entire collection of general genetic data currently available for that species24,25. Our laboratory has also developed mass spectrometry-based technologies for targeted and non-targeted metabolomic analysis26. By implementing these background and novel methods, we hope to provide robust evidence upon which to evaluate any relationship between the JA pathway and metabolic biosynthesis in S. miltiorrhiza. As part of our first step, we report here our comprehensive analysis of the impact that MeJA has on S. miltiorrhiza along with a phenotype description, transcriptome data and results from the metabolomic analysis. In this study, we also selected a candidate gene induced by MeJA that encodes JAZ protein. This gene, SmJAZ8, was identified as a repressor in the JA signaling pathway. Its abundance of transcript was linked with extraordinary suppression of the regulation of JA-dependent responses in S. miltiorrhiza. Transgenic analysis also suggested its potential role in JAZ-regulated secondary metabolism, based on significantly increased levels of salvianolic acid B (Sal B) measured in the transgenics. These findings will help in further establishing an efficient strategy as “phenotype-transcriptome-metabolome” for discovering novel genes in those economically significant medicinal plants.
Results
“Phenotype-metabolome-transcriptome” analysis of Salvia miltiorrhiza under MeJA treatment
Plants exposed to MeJA also displayed massive, spontaneous necrotic lesions on leaves nearest the base of the plant. This necrosis eventually engulfed the entire surface of older leaves so that they became dark and died by Day 14 (Fig. 1A). It has been known for a long time that root growth is inhibited by JA or its methyl ester27. To investigate the effects of MeJA in S. miltiorrhiza, we initially cultured plants for 3 d in an MS basal solid medium before exposing them to either 100 μM MeJA or a mock solution. A scan showed that roots harvested from JA-treated seedlings were shorter and had a significantly slower growth rate when compared with the phenotype of roots from control plants (Fig. 1B). On Day 14 of these observations, the treated plants exhibited a moderate increment in average root diameters but significantly fewer (by 90%) lateral roots (Fig. 1C, Table S1).

Response by Salvia miltiorrhiza to MeJA.
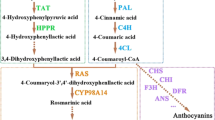
Phenotype of shoots (A) and roots (B) upon treatment. (C) Effects of MeJA on root growth relative to MOCK control. (D) Map of major metabolic pathways relative to secondary metabolaism of S. miltiorrhiz. Red font indicates that metabolite level was changed by more than 1.5-fold (p < 0.05) when compared with untreated control; black, no significant changes; gray, undetectable. Time course of MeJA effects on contents of rosmarinic acid, RA (E) and salvianolic acid, SalB (F) in MOCK seedlings under 0.2% ethanol treatment and in MeJA seedlings under 0.1 mM treatment. *significant at p < 0.05; **highly significant at p < 0.01. (G) proposed biosynthetic pathway for phenolics, beginning with core phenylpropanoid and tyrosine pathways and leading to 3 major branch pathways: phenolic acid, flavonoids and lignin (up-regulated genes in red; down-regulated genes in green). ANS, anthocyanin synthase; CAD, cinnamyl alcohol dehydrogenase; CCR, cinnamoyl-CoA reductase; CCoAMT, caffeoyl-CoA O-methyltransferase; CHI, chalcone isomerase; CHS, chalcone synthase; COMT, caffeic acid O-methyltransferase; CYP, cytochrome P450enzymes; CPR, cytochrome P450 reductase; C3′H, coumarate 3′-hydroxylase; C4H, cinnamate 4-hydroxylase; DFR, dihydroflavonol 4-reductase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; F5H, ferulate 5-hydroxylase; FLS, flavonol synthase; HCT, hydroxyl cinnamoyl transferase; HPPR, hydroxyl phenylpyruvate reductase; LAR, leucocy anidin reductase; PAL, phenylalanine ammonia lyase; RAS, rosmarinic acid synthase; TAT, tyrosine aminotransferase; 4CL, hydroxycinnamate-CoA ligase. (H) MeJA signal pathway. (I), qRT-PCR verification of DGE tag data for JAZ (Unigene36977); F3H (Unigene54426); TAT1 (Unigene56449); CAD (Unigene37246); CCR1 (Unigene53802); CCoAMT (Unigene39895); RAS (Unigene55331); 4CL1 (Unigene54261); C4H (Unigene53935) and PAL2 (Unigene52119). All data are means of 3 replicates, with error bars indicating SD.
We suspected that, to a large extent, this influence on the morphological phenotype depended upon modulations in primary metabolism. Therefore, we performed GC/MS-based profiling for primary metabolites as well as phenolic- and tanshinone-targeted analyses by LC/MS.
As expected, we identified 16 primary metabolites as differential contributors (Table S2). These included amino acids (threonine, proline, asparagine and glutamine), carbohydrates (D-fructose, D-ribose, glucose oxime, D-mannitol, mannitol, galactose oxime, sucrose and lactose), organic acids (malic acid and butanoic acid) and fatty acids (palmitic acid and stearic acid). Those compounds strongly influenced plant growth. In addition, the levels of secondary metabolites were determined with LC-MS (Table S3) and a total of 19 compounds were differentially accumulated between the two groups.
In all, we extracted 132 and 125 independent analytes that represented putative metabolites obtained from the LC/MS and GC/MS data sets, respectively. After peak alignments, both sets were assessed by the unsupervised PCA and PLS-DA method. Differences between the two groups were distinct (Figure S2). When combined with our finding from the score and loading plots (Figure S3), these results suggested that both primary and secondary metabolite contents had been altered by MeJA stimulus (Fig. 1D).
It is widely believed that rosmarinic acid (RA) and salvianolic acid B (Sal B) are the active phenolic ingredients in S. miltiorrhiza8,28. Therefore, we selected RA and Sal B as the representative compounds for further investigation. Their accumulations in response to MeJA were monitored over 0, 3, 6, 9 and 14 days, respectively. Duplicate analyses were conducted via HPLC to develop a time course of reactions (Fig. 1E,F). Levels of RA increased up to Day 3, peaking at 29.68 ± 0.09 mg g−1 DW when compared with the corresponding control. The accumulation of Sal B was also highest on Day 3 before declining. Previous research has shown that accumulations of RA and Sal B in S. miltiorrhiza are highest when exposed to 100 μM MeJA for 6 d and that those levels are correlated with increments in gene expression6. Hence, we concluded that these enhanced accumulations were probably a result of increased levels of transcripts for genes involved in those metabolic pathways.
Using the Solexa/Illumina DGE system, we found a total of 3,843,001 and 3,787,000 tags in our control and MeJA libraries. After low-quality tags were filtered out, 3,819,543 and 3,763,892 “clean” tags remained, respectively. These were mapped to the previously reported reference library24 for bioinformatics purposes. Approximately 48% of all clean tags could be mapped to >25% of the predicted S. miltiorrhiza genes, i.e., 25,251 and 23,298 genes for the control and MeJA libraries, respectively (Table S4). Overall, 0.82% of these genes were significantly up-regulated by more than 5-fold, whereas the expression of 1.05% of all genes was decreased in the MeJA library (FDR ≤ 0.001 and |log2Ratio|≥1; Figures S4, S5). We used PCA to compare the transcript profiles between MOCK and MeJA-treated plants. The differences in those profiles were similar to the contrasts found in metabolite profiles between the two libraries (Figure S2, S3). The KEGG pathway was analyzed to improve our understanding of DEG biological functions and to identify genes potentially involved in crosstalk between the JA signaling pathway and important biochemical pathways. Overall, we found that our DEGs participated in 116 pathways, with 24 being significantly enriched in DEGs at p < 0.05 and an FDR value < 0.01 (Table S5). Those functions included the metabolism of phenylpropanoids and alkaloids, as well as the biosynthesis of hormones, phenylpropanoids and flavonoids. Based on previous experiments conducted in our laboratory, we chose to focus on genes in the pathways for phenolic acids and mapped them with KEGG pathway annotations to predict candidate genes important for JA-induced metabolism. Phenolic compounds are produced from phenylalanine and tyrosine through a series of hydroxylation, methylation and dehydration reactions (Fig. 1G). Expression of 8 genes in the phenylpropanoid pathway increased more than 2-fold, especially PAL, 4CL, RAS, CYP98A14 and CPR (Table S6, Fig. 1G). This discovery was consistent with an earlier report of JA-induced expression in Larix gmelinii29. We detected three genes in the tyrosine pathway, where expression was enhanced by 3.10-fold to 5.57-fold over the control. Other genes were involved in the phenylpropanoid pathway to regulate the production of lignin and flavonoids30,31. We noted that the abundances of several transcripts in those two pathways were elevated after plants were exposed to MeJA whereas expression of nine genes was reduced. We also found it of interest that some of the unigenes annotated in same gene shown different expression patterns and vice versa. For example, expression of Unigene2612 was down-regulated while that of 56687 was up-regulated simultaneously, even though both were annotated by flavanone 3′-hydroxylase (F3′H).
In addition to analyzing the abundance of genes for enzymes in the biosynthesis pathway for phenolics, we determined whether upstream changes in the JA signal pathway are responsible for the extraordinary accumulation of metabolites in MeJA-treated plants. As described previously, JA biosynthesis is regulated by a positive feedback loop32. Of those genes, most had higher transcripts under MeJA treatment (Fig. 1H). In particular, LOX expression was increased by up to 133.00-fold when compared with the control.
Jasmonoyl isoleucine conjugate synthase (JAR) catalyzes the final step in the formation of the bioactive JA compound27. That gene was the only one in the JA biosynthesis pathway that was significantly suppressed. Such a cycle of expression may have contributed to the fine-tuning of JA signaling. Similar to previous reports33, we found that the transcripts of five unigenes annotated as JAZ were up to 98.12-fold more abundant in MeJA-treated plants (Table S6). Due to the core role of JAZ protein in the JA-mediated metabolic reprogramming, we thus focused on those ‘pop’ genes and analyzed their functions in details.
Eight up-regulated and two down-regulated genes were randomly selected as representatives within different pathways. Pair-wise comparisons of the normalized abundance of these 10 genes were made using results from our DGE and qRT-PCR analyses. These revealed similar patterns of induction by MeJA (Fig. 1I), which confirmed the reliability of our transcriptome data.
Identification of novel S. miltiorrhiza JAZ genes
Conserved domains for ZIM and Jas were utilized to screen sequence fragments from our transcriptome database24 so that we could identify putative JAZs from S. miltiorrhiza. Based on Blast results, we designed primers flanking coding regions that resembled other known JAZs. Using leaf- and/or root-derived cDNAs, we cloned and sequenced PCR-amplified fragments, thereby yielding four distinct genes named SmJAZ1, -2, -3 and -8 (corresponding GenBank Accession Numbers JQ936590, KC864779, KC864780 and JQ936591).
The expression patterns of SmJAZs were studied with qRT-PCR as well as their response to stress. As our control, expression was monitored in untreated leaves to understand systemic induction. Whereas SmJAZ1, -2 and -8 were mainly expressed in leaf and stem tissues, only SmJAZ3 was more highly expressed in the roots (Fig. 2). Similar to previous reports33, all SmJAZs showed strong and early induction by MeJA or wounding. GA treatment led to immediate down-regulation while PAC, an inhibitor of GA, caused SmJAZs to be moderately up-regulated. These patterns were similar to those found with other species, which directly confirmed the universal characteristics of JAZ genes and suggested that their functions are conserved in hormone and defense pathways34. The response to wounding or MeJA was quite different between SmJAZ8 and the other SmJAZs, with transcripts of the former being more than 100-fold (wounding) and 17-fold (MeJA) higher at 0.5 h after elicitation (see scales along y-axes in Fig. 2). This suggested that, compared with its lack of detection under control conditions, SmJAZ8 has special biological functions and is inducible only by certain stress stimuli.

Expression patterns of individual JAZ genes in different tissues and under abiotic stresses.
(A–D) Transcript abundances for SmJAZ1 (A), SmJAZ2 (B), SmJAZ3 (C) and SmJAZ8 (D) in root, stem, leaf and flower. (E–H) Expression following treatment with 5 mM MeJA (E), mechanical wounding (F), 150 μM GA (G) and 150 μM PAC (H). Accumulations of SmJAZ transcripts were determined by qRT-PCR (n = 3). β-Actin was used as internal control. Fold-changes indicate expression relative to negative control, which was set to 1. Significant differences were determined at each time point compared with negative control, by Student’s t-tests (*P < 0.05, **P < 0.01, ***P < 0.001).
A detailed structure analysis of SmJAZ8 showed that it has three exons and two introns. Sequence alignments showed that, like other JAZ family members, SmJAZ8 contains the ZIM and Jas domains. Phylogenetic analysis of the full-length proteins from S. miltiorrhiza and members in the Arabidopsis JAZ family33 (AtJAZ1 to -12) showed that SmJAZ genes could be clustered with others in Subgroup III because it was most similar to AtJAZ7 and AtJAZ8 (Figure S6)5.
Overexpression of SmJAZ8 and phenotype analysis of jasmonate-responsiveness
To characterize the functioning of SmJAZ8 in detail, we generated transgenic plants that over-expressed SmJAZ8 (Figure S7). Our qRT-PCR analyses of expression in PCR-positive transgenics showed that SmJAZ8 transcripts were accumulated by more than 2000-fold over the controls. Four lines exhibiting efficient overexpression were selected for functional analysis. However, the transgenic sterile plants were no difference from the controls in vegetative growth and development under normal conditions.
To evaluate the role of SmJAZ8 in the JA signaling pathway, we monitored the expression of inducible genes SmC4H, SmTAT, SmRAS and SmF3H (based on DGE results above) in OEB and control plants following MeJA exposure. Their transcripts in the control lines were substantially induced early while none of those genes was induced in any transgenic line within the first 3 h of treatment (Fig. 3). The OEB plants seemed to be JA-hyposensitive presumably because of their high level of SmJAZ8 expression.

Patterns of expression by JA-responsive genes in OEB lines under MeJA treatment.
Mean values and SDs were obtained from 3 technical and 3 biological replicates.
The inhibition of root growth by exogenous JA has long been used to screen for JA-insensitive mutants such as the coi1 mutant35. Jasmonate is also reportedly involved in the formation of lateral roots36. Because SmJAZ8 likely represses MeJA-induced genes, its overexpression should result in weak constitutive physiological responses, e.g., reductions in root growth or development of lateral roots. When our three-day-old seedlings received exogenous MeJA, the root growth of MeJA-treated control group was obviously inhibited when compared with either untreated plants or treated OEB lines. Statistical analysis of those data also confirmed that this overexpression of SmJAZ8 buffered the influence of MeJA on lateral root formation (Table 1).
In S. miltiorrhiza, the predominant active ingredients, RA and Sal B, are enriched by MeJA. To examine whether this is consistent with the transgenics, we used HPLC to measure their accumulations after 14 d of treatment. Whereas differences in Sal B contents did not differ noticeably between control and overexpressing plants (except for Line OEB-5), levels of RA were significantly lower in the OEB lines (Table 2). Namely, the JA-induced metabolite increments seemed to be reduced partially in transgenic plants.
Levels of total phenolics and flavonoids and antioxidant properties of plant extracts
Although SmJAZ8 overexpression did not affect plant growth and development, the amounts of less-essential secondary metabolites may have been altered. Therefore, we performed global assays for phenolics and flavonoids. In contrast to our expectations, the levels of both were higher in OEB lines than in the controls. In particular, the strongest increments in constitutive levels were found in OEB-4, i.e., 2.45-fold higher for total phenolics and 1.75-fold higher for flavonoids (Fig. 4A,B).

Changes in targeted secondary metabolites and antioxidant capacity between control and OEB lines.
Levels of total phenolics (A) and flavonoids (B) from root extracts. (C) curve for DPPH free radical scavenging. (D) Trolox equivalent antioxidant capacity. Values followed by **within a column are significantly different from the control at p < 0.01.
Secondary metabolites, especially phenolic compounds, may have bioactive properties and be important contributors to antioxidant, antimicrobial and anti-inflammatory activities37,38. Thus, the over-accumulation of flavonoids may enhance crop tolerances to both biotic and abiotic stresses39. Here, the antioxidant capacity of our transgenic plants was obviously enhanced (Fig. 4C,D). Calculations revealed that the EC50 value in OEB-4 was 0.237 mg mL−1, which was significantly higher than the 0.537 mg mL−1 measured for the control. In contrast, none of the samples showed activity as strong as VC. These results suggested that overexpressing plants had a higher level of secondary metabolites that was most likely responsible for their greater antioxidant activities.
Variations in specific soluble phenolic acids
We performed HPLC analysis to study the levels of RA and Sal B, two major hydrophilic and active pharmaceutical ingredients in S. miltiorrhiza. We also investigated how their metabolism was modified in OEB plants. Concentrations were determined at 30 d (just after plants were transferred to the greenhouse) and again at 150 d. The Sal B concentration was highest (78.74 ± 1.48 mg g−1 DW) in OEB-4 roots sampled at 150 d and was 1.41-fold higher then, as well as 1.30-fold higher on Day 30, than corresponding levels in the control (Fig. 5). Similarly, amounts of RA were significantly higher (by 1.52-fold to 2.15-fold) in one-month-old OEB lines. However, those levels began to decline after 150 d. Overall, the status of Sal B was more stable than that of RA, presumably due to that Sal B is the final product in the phenolic acid pathway.

Specific changes in concentrations of RA (A) and Sal B (B) in roots from OEB and control lines, based on HPLC results.
(C) verification of relative gene expression via qRT-PCR. Fold-changes reflect expression in OEB-4 and OEB-7 compared with control expression, for which values were set to 1 (not shown).
To evaluate whether these substantially elevated accumulations of RA and Sal B in the transgenics were a result of greater gene expression relative to the control, we examined transcript levels for nine key structural genes in the phenolic biosynthesis pathway as well as three other SmJAZs. When normalized to the control, those three SmJAZs were shown to be down-regulated simultaneously by 19 to 32% (Fig. 5). Expression of PAL, hydroxyl phenylpyruvate reductase (HPPR), TAT and RAS, which are required for RA synthesis, was up-regulated in OEB plants. We observed no distinct increases in the transcripts of hydroxyl cinnamoyl transferase (HCT), chalcone synthase (CHS) and F3H, all of which are involved in pathways for flavonoid and lignin biosynthesis. Transcripts of dihydroflavonol 4-reductase (DFR) were higher only in OEB-7. Those results were consistent with findings from our previous study40, in which Sal B concentrations were enhanced when gene expression increased in the biosynthesis pathway.
Discussion
The power of integrating transcriptome and metabolome data for functional genomics
In the process of discovering two glycosyltransferase genes putatively involved in anthocyanin synthesis, researchers recognized that an approach combining metabolomics and transcriptomics can be an efficient means for functional classification of genes21. To complete their examination of how enzyme genes and the pathways for flavonoid metabolism are modified, Keiko et al.30. identified several novel genes from a pool of candidates by coupling their detailed analysis of secondary metabolites with transcriptome coexpression analysis. Those reports provided strong evidence that integrating transcriptome and metabolome data can be applied to studies of roles by hypersensitive response-associated genes in tomato41. By combining metabolomic and transcriptomic analysis, terpenoid metabolism were demonstrated to display a biphasic response to elicitation23 and a potential role for the induced MeJA-responsive transcription factor was found. However, studies focused on the phenolic acid pathways are still lacking. Transcriptional profile of S. miltiorrhiza leaves in response to MeJA induction was detected by RNA-seq technology and the results shown a significant transcriptional complexity20. Subsequently, the same group described 29 genes related to phenolic acid bioynthesis from the S. miltiorrhiza genome10 and provided a rich gene pool for phenolic acid biosynthesis. The extensive functional evidences and their responses to JA induction are of great importance. Here, we used these methods to reveal the holistic changes associated with exogenous MeJA. Massive re-programming occurred in our JA-treated plants and we found DEGs, most of which were up-regulated, that belonged to 116 pathways.
Crosstalk between the JA signal pathway and the secondary metabolism pathway is one of the best examples of cooperation among genes to fulfill their biological functions. Because MeJA treatment was linked with increased transcript abundance, it also led to a marked enrichment in levels of primary and secondary metabolites. For example, two active ingredients, RA and Sal B, were accumulated in concentrations much above normal levels. Such results can improve our understanding of the roles played by individual components in the JA response and these findings help to explain how the functional genes examined here contribute to this metabolomic network.
Conserved functioning of SmJAZ8, a novel JAZ gene in S. miltiorrhiza
The JAZ proteins are targets of the SCFCOI1 complex; their degradation releases some positively acting TFs to initiate transcription of JA-responsive genes42. However, Thines et al.15 have produced transgenic lines in Arabidopsis with complementary DNAs that encode JAZ proteins under the CaMV 35S promoter. None of those plants exhibit an obvious jasmonate-related phenotype. Other reports have indicated that only overexpression or a reduction in the transcript abundance of alternative splice variants of AtJAZ10 (i.e., JAS1, At5g13220) affects plant growth in response to either MeJA treatment or wounding16. Furthermore, Shyu et al.43 have shown that 35S:AtJAZ8 plants have greatly increased capacity to repress JA responses. This suggests that the LPIARR motif is an important signal of degradation, but is lacking in AtJAZ8, which leads to greater protein stability. Here, those unique features were also manifested by SmJAZ8. When over-expressed, its transcript repressed the ability of MeJA to inhibit root growth and blocked the activation of JA-inducible genes. This is probably because, as with AtJAZ8, its structure does not contain the LPIARR motif.
Our results also showed that modulating the levels of SmJAZ8 transcript partially altered the MeJA-responsive accumulation of phenolic acids, which indicated that the JA-induced upregulation of RA and Sal B is mediated by this JAZ repressor.
Stress-induced expression of SmJAZ8 may prevent excessive activation of JA responses in plants, similar to how AtJAZ8 and the alternative splice variant of AtJAZ10 participate in negative feedback control of jasmonate signaling44. Therefore, we propose that SmJAZ8 acts as a repressor that can strongly affect the downstream response to this signal.
The involvement of SmJAZ8 in biosynthesis of phenolic acids
One of our goals was to understand the crosstalk between the JA signal pathway and production of phenolic acids. Specifically, we wondered whether blocking transduction, either entirely or partially, of JA signal could alter the accumulation of phenolic metabolites in S. miltiorrhiza. In tobacco, only the expression of dominant-negative truncated JAZ constructs can interrupt the COI1-JAZ pathway and depress basal levels of nicotine18. In JAZ1∆3A-transformed Arabidopsis, which constitutively expresses truncated AtJAZ1 that lacks the C-terminal Jas-containing region, much less anthocyanin is accumulated following MeJA treatment, while no remarkable differences in levels are measured under normal growing conditions19. A common feature underlying JA-regulated biosynthesis of secondary metabolites is the involvement of JAZ proteins along with specific TFs and other components. Further research is needed to elucidate the distinct pathways by which diverse metabolites are activated in different plant species45. Here, biosynthesis of total phenolic acids and flavonoids was enhanced in transgenic plants and antioxidant capacity was also increased. Consistent with a role for JAZ in forming homo- and heterodimers46, we found that expression was decreased for some JAZ genes, including SmJAZ1, SmJAZ2 and SmJAZ3, which indicated that other pathways exist. As we had predicted, basal expression of PAL, HPPR, TAT and RAS was up-regulated while Sal B accumulated to higher levels in OEB-transgenic plants.
All of our results suggest that crosstalk is possible among individual JAZ proteins, as illustrated by the positive regulatory effect of the SmJAZ8 repressor on phenolic acid accumulations. Furthermore, the high production of secondary metabolites may led to attenuate the JA responses by stably expressed SmJAZ8 and induction of its expression in wild-type plants was strongly activated by MeJA from much lower basal levels. Therefore, we propose that these associated metabolite accumulations occur downstream of SmJAZ8 activation. While the extent of the contribution by this gene to that phenomenon is unclear, it may be part of other pathways. Similar crosstalk within JAZ-regulated secondary metabolism has been described for Nicotiana attenuata, where the silencing of NaJAZh significantly reduces nicotine levels47. Furthermore, overexpression of GsJAZ2 in Glycine soja leads to enhance plant tolerance with positive regulation of expression of stress-inducible marker genes48. Thus, it should also be possible to identify the differentially expressed TFs that are directly integrated with SmJAZ8 or other JAZ proteins to regulate secondary metabolism in S. miltiorrhiza. For example, R2R3-MYB factors are involved in modulating many phenolic pathways49, such as AtPAP1, which induces the accumulation of anthocyanin in Arabidopsis19 and Sal B in S. miltiorrhiza40. Because of this, it is tempting to speculate on the existence of a PAP1 in S. miltiorrhiza that is controlled by JAZ protein and which could control phenolic metabolism. Future investigations of any relationship between PAP1 and JAZ genes in S. miltiorrhiza should improve our understanding about the novel crosstalk in JAZ-regulated secondary metabolism within this model medicinal plant.
Materials and Methods
Plant materials and treatments
Seeds of Salvia miltiorrhiza Bunge, collected from Shangluo, Shaanxi Province, China, were germinated in vermiculite. When their fourth true leaves appeared, the plantlets were transferred to pots filled with soil (two per pot), then grown in our laboratory’s greenhouse. Temperatures ranged from 24 to 26 °C under a natural. Plants were irrigated with distilled water at 3-d intervals. Prior to the treatment period, all plants were acclimated for one week in a growth chamber under a 16-h photoperiod provided from cool-white fluorescent lamps (25 μmol m−2 s−1). The six-month-old plants were then randomly assigned to treatment groups. To examine the effect of MeJA (Sigma Chemical Co.), we sprayed the plants with either a 500 μM MeJA solution (100 mL per plantlet) that was mixed with 2% ethanol (v/v) in water (treatment solution) or else a mock (MOCK control) solution that contained only the ethanol/water mixture. After the plants were hermetically sealed in plastic bags for 1 h, they were exposed to open air for 2 h so that any residual ethanol water and MeJA could evaporate from the plants. Samples for transcript profiling were harvested from three biological replicates at 6 h after treatment, then immediately frozen in liquid nitrogen and stored at −80 °C. For metabolite profiling, plantlets from these two treatment groups were harvested at 0, 3, 6 and 12 d, then dried at 30 °C before being pulverized to a fine powder (2-mm mesh) and stored at 4 °C.
To ensure stable asexual reproduction, we surface-sterilized S. miltiorrhiza seeds with a 1% (v/v) mercuric chloride solution for 8 min and with 75% (v/v) ethanol for 25 s. They were then thoroughly washed in sterile water and placed for 7 d at 25 °C in the dark on an MS basal medium containing 0.65% agar. Uniformly germinated seeds were selected and cultured on a fresh MS basal medium containing 0.65% agar and 3% sucrose, as described by Yan and Wang50. The MeJA treatment described above was repeated with these sterile plantlets to verify those results. After approximately one month of growth, two leaf blades at the top of each selected stem were removed and placed on another solid MS-based medium via the clipping inoculation technique. These seedlings were then grown for another 3 d before being transferred to a medium supplemented with either MeJA (100 μM final concentration) or the mock solution (ethanol/water mixture). For quantitative real-time PCR (qRT-PCR) analysis, seedlings were harvested at Hour 6 and frozen at −80 °C. Contents of secondary metabolites were monitored over time by HPLC. Root lengths were measured on Day 14, followed by washing with distilled water and scanning at a resolution of 600 dpi. The root phenotypes were examined with WinRHIZO software (Aozuo, China).
Hormone treatments were conducted by spraying the leaves of three-month-old seedlings with 5 mM MeJA, 150 μM gibberellin (GA3), or 150 μM paclobutrazol (PAC). Control seedlings received only water. Tissues were sampled after 0, 0.5, 1, 2, 4, 6, 8, 12 and 24 h.
To investigate the effect of mechanical wounding, we crushed the leaf midribs three times with a hemostat, using approximately four leaves per plant to damage approximately 40% of the total area33. Samples were collected for analysis after 0, 0.5, 1, 2, 4 and 6 h.
All experiments were repeated at least three times; results from one representative experiment are presented here.
Transcript profiling
To identify the genes associated with the JA signaling pathway in S. miltiorrhiza, we used Solexa sequencing to perform digital gene expression (DGE) profile analysis of our treated plants at Day 180. Total RNA was isolated from each sample with RNeasy Plant Mini kits (OMEGA, China) according to the manufacturer’s instructions. The quality and concentration of total RNA were measured by 1.0% agarose gel electrophoresis and spectrophotometric analysis (SHIMADZU UV-2450). The RNA samples >300 ng μL−1 with high purity (OD260/280 > 1.8, OD260/230 > 1.5) underwent transcriptome analysis and qRT-PCR. Digital expression and sequencing were performed at BGI in Shenzhen, China (http://www.genomics.org.cn/). Sequences were compared by conducting a Blastn search with two databases: NCBI and our transcriptome for S. miltiorrhiza24. All clean tags obtained by filtering out any adaptor-only tags and low-quality tags were annotated based on S. miltiorrhiza reference genes (Table S4). For statistical analysis, we used a method previously described by Zhang et al.26 to identify differentially expressed genes (DEGs) based on false discovery rates (≤0.001), p-values of < 0.005 and estimated absolute log2 fold-changes of ≥1 in sequence counts across libraries (Figures S4, S5). Significantly enriched metabolic pathways or signal transduction pathways were functionally classified into several categories with KEGG (http://www.genome.jp/kegg/) by comparing those DEGs with the control genome background (Table S5). Pathways with Q values of < 0.05 were considered to be significantly enriched in DEGs.
Metabolite profiling by GC/MS and LC/MS
All samples were harvested and extracted as described previously by Zhang et al.26 Air-dried roots from treated and untreated samples (60 mg each) were extracted, derivatized and prepared for GC/MS using a scaled-down version of a two-phase methanol/chloroform method51. For profiling, 1 μL of supernatant was applied to an Agilent 6890N GC-FID and Shimadzu QP 2010 GC/MS (Kyoto, Japan), using a 20:1 split injection ratio. Separations were made on a fused silica capillary Agilent Technology HP-5ms (5% phenyl methyl siloxane) column (30 m × 0.25 mm i.d., 0.25 μm film thickness). The injector and detector temperatures were 250 °C and 200 °C, respectively. The oven temperature was 136 °C for the first 5 min, but was then ramped to 300 °C in 4 °C increments, with a Nitrogen gas linear velocity of 1.56 mL min−1 and a data acquisition rate of 20 Hz. Full-scan mass spectra were acquired from 40 to 600 m/z. Mass spectrometry was performed at source and interface temperatures of 200 °C and 250 °C, respectively, with a voltage of 0.9 kV. Metabolites were identified according to the protocol of Ma et al.52, which had been adapted by Zhang et al.26 for use with S. miltiorrhiza. We subsequently obtained a peak table of whole samples, including retention times and MS fragment ions (Table S2), by comparing their mass spectra with those from the National Institute of Standards and Technology(NIST, USA) library as well as with standard compounds. No peak was available from the table if its area was zero in more than 80% of the samples. The relative areas of the remaining 16 peaks were mean-centered (Ctr) or pareto-scaled (Par), then evaluated by Principal Component Analysis (PCA) and partial least squares-discriminant analysis (PLS-DA), via the SIMCA-P software program (Umetrics, Umea, Sweden). Independent one-way ANOVAs were used with SPSS (version 13.0) to select variables with VIP (variable importance in the projection, more than 1) p-values of < 0.05. These were chosen because we believed any significant differences in compounds between our control and treated groups were evidence of important roles in functional classifications.
For LC/MS, air-dried samples were homogenized in 500 μL of extraction solvent (3:1 methanol:H2O) per 100.0 mg of dry weight. The extracting procedure was performed as described by Zhang et al.40. Afterward, 20 μL of supernatant was applied to an Agilent 1100 HPLC system (Agilent Technologies, Palo Alto, CA, USA) that comprised a quaternary solvent delivery system, on-line degasser, auto-sampler, column temperature controller and photodiode array detector coupled with an analytical workstation. HPLC was performed on a Phenomenex C18 column (5 μm, 250 mm × 4.6 mm) at a flow rate of 1.0 mL min−1 (temperature 30 °C). The elution gradient included Solvent A (0.4% acetic acid in water), Solvent B (acetonitrile) and Solvent C (methanol) and followed a profile of 0 min, 95% A, 5% B, 0% C; 5 min, 90% A, 10% B; 25 min, 67% A, 30% B, 3% C; 40 min, 60% A, 35% B, 5% C; 55 min, 49% A, 45% B, 6% C; 60 min, 47% A, 45% B, 8% C; 80 min, 35% A, 55% B, 10% C; 90 min, 25% A, 60% B, 15% C; 95 min, 10% A, 70% B, 20% C; and 100 min, 80% B, 20% C). Linear gradients were used between time points. A PDA was utilized for detecting UV-visible absorption between 250 and 650 nm, with samples measured at 280 nm. Mass spectra were acquired by a Bruker Esquire 6000 ion trap instrument with an ESI source (Bruker Daltonics, Inc., USA). For phenolic acids, the ion chromatograms of negative electrospray ionization were recorded from 0 to 60 min by a mass spectrum detector. The spectrum was scanned and stored from m/z of 100 to 1000. For the tanshinones, the ionization mode was positive (60 ~ 100 min) and spectra were recorded between 100 and 800 m/z. The drying gas flow was 12 L min−1 and the nebulizer pressure was set to 35 psi at a capillary temperature and voltage of 320 °C and 30 V, respectively. The collision-induced dissociation (CID) energy was adjusted to 40 ~ 50%. Metabolites were identified based on their retention times, UV spectra and mass fragmentation by applying tandem MS analysis and comparing results with known compounds in our laboratory data26. As authentic standards, caffeic acid, rosmarinic acid, salvianolic acid B, cryptotanshinone, tanshinone I and tanshinone IIA were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China).
Quantitative real-time PCR analysis
To evaluate the reliability of DEGs identified by DGE, we used qRT-PCR to profile candidate genes that might respond to MeJA treatment. Based on the KEGG pathway annotation, 10 genes were selected randomly to compare their expression patterns under different conditions. The cDNA was generated from total RNA using a Revert Aid First Strand cDNA Synthesis Kit (Takara) according to the manufacturer’s instructions. We chose Actin as the internal reference gene for normalization because its expression has previously been proven stable in S. miltiorrhiza53. Relative gene expression was evaluated by using Real Master Mix SYBR Green II dye (Takara, Japan) and the iQ™ 5 Real-time PCR detection system (Bio-Rad, USA), following earlier protocols40. The primers used for detection are listed in Table S7.
Contents of targeted phenolic compounds determined by HPLC
We targeted two phenolic compounds to examine the effects of MeJA on accumulations of secondary metabolites. Seedlings exposed to MeJA treatment for 0, 3, 6, 9, or 12 d were sampled and ground into powders, which were then extracted as described above. The short (40-min) procedure for HPLC analysis was the same as for metabolite profiling.
Isolation and sequence analysis of SmJAZs
To identify members of the JAZ gene family within S. miltiorrhiza, we screened the DGE and transcriptome databases that had been constructed and conserved by our laboratory24. Sequences were then assembled into putative JAZ genes by using DNAStar SeqMan 5.0 software. Those assemblies were compared against the nonredundant peptide database at the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). The performance of four fragments was more similar to other JAZ genes from Arabidopsis thaliana and contained two conserved ZIM and JAZ motifs. Primers were designed (Table S7) to clone the corresponding sequences. If necessary, the primers were redesigned until a clear PCR product of estimated length could be amplified from the cDNA template. All PCR products were purified and cloned into a pGEM-T easy vector (Promega, USA) before sequencing. At least three independent clones were fully sequenced for each type of insert.
To test expression in response to hormones, we also designed gene-specific primer pairs (Table S7). Total RNA was also extracted from the roots, stems, leaves and flowers of two-year-old plants to investigate transcription patterns. That age was used so that we could obtain flower organs. All reactions were performed under the qRT-PCR conditions described above.
The sequence of SmJAZ8, a novel JAZ gene, was also amplified by PCR, using total DNA as template. Genomic DNA was isolated from the leaves of three-month-old seedlings per the modified cetyl-trimethyl ammonium bromide method54. Combined with the cDNA sequence, a diagram of exon/intron structures was obtained using public online software from the Gene Structure Display Server (http://gsds.cbi.pku.edu.cn), which exhibited both exon positions and conserved domains. The full-length sequence of its translated putative protein was aligned with those of other JAZ proteins by the ClustalW program and a phylogenetic tree was built using bioinformatics software MEGA 5.0 (Figure S6), the maximum parsimony (MP) method and a bootstrap test (1,000 replicates).
Plant transformation
To generate transgenic plants, we amplified the coding regions of SmJAZ8 from pGEM-T-SmJAZ8 (described above). The sequence was inserted into the pKANNIBAL vector, placed under the CaMV 35S promoter. The construct was restricted by Not I and the digested fragment was ligated to the pART-27 vector. The final recombined plasmid was introduced into Agrobacterium tumefaciens EHA105 and transgenic S. miltiorrhiza plants were generated by the leaf-disc method.50 Transformation was verified by screening with kanamycin and cephalosporin. Our controls were either wild-type plants or those transformed with only the binary vector pKannibal-pART27. Transgenic lines were confirmed by PCR screening using genomic DNA and qRT-PCR was used to select the best overexpression transgenic lines — OEB-2, OEB-4, OEB-5 and OEB-7. One-month-old plants were used for preliminary quantification of phenolic acids as well as investigations of total flavonoids and the MeJA-responsive phenotype. For HPLC analysis of the phenolic compounds, those transgenics and the control plants were transferred into soil and grown in a greenhouse for at least one month.
Analysis of total phenolics, total flavonoids and antioxidant capacity
The roots from one-month-old plants were air-dried at room temperature. The methodology for extracting and determining total phenolics and flavonoids followed procedures described by Zhang et al.40.
Total antioxidant activities were evaluated for trolox equivalent antioxidant capacity (TEAC), based on our previously published protocols26.
For assaying the scavenging of free radicals in methanolic extracts, we utilized the DPPH method of Zhang et al.55 with slight modifications. Different dilutions of the extracts (2 mL; 0.1–1.5 mg mL−1) were added to 1 mL of a 0.004% methanol solution of DPPH. The positive reference was vitamin C (VC). After 30 min of incubation at room temperature, scavenging activity was spectrophotometrically measured at 517 nm and was calculated by the following formula:

where Ablank was the absorbance of the control reaction and Asample was the absorbance of the test sample.
The EC50 value was used to represent the antioxidant capacity of the crude extracts and VC standard, i.e., the effective concentration at which DPPH free radicals were depleted by 50%.
Additional Information
How to cite this article: Ge, Q. et al. Combination of transcriptomic and metabolomic analyses reveals a JAZ repressor in the jasmonate signaling pathway of Salvia miltiorrhiza. Sci. Rep. 5, 14048; doi: 10.1038/srep14048 (2015).
References
Ma, Y. et al. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J Exp Bot 63, 2809–2823 (2012).
Xu, Z. et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J 82, 951–961 (2015).
Han, J. Y. et al. Ameliorating effects of compounds derived from Salvia miltiorrhiza root extract on microcirculatory disturbance and target organ injury by ischemia and reperfusion. Pharmacol Ther 117, 280–295 (2008).
Zhao, J. L., Zhou, L. G. & Wu, J. Y. Effects of biotic and abiotic elicitors on cell growth and tanshinone accumulation in Salvia miltiorrhiza cell cultures. Appl Microbiol Biotechnol 87, 137–144 (2010).
Yang, D. L. et al. Plant hormone jasmonate prioritizes defense over growth by interfering with gibberellin signaling cascade. Proc Natl Acad Sci USA 109, E1192–1200 (2012).
Xiao, Y. et al. Methyl jasmonate dramatically enhances the accumulation of phenolic acids in Salvia miltiorrhiza hairy root cultures. Physiol Plant 137, 1–9 (2009).
Gu, X. C. et al. Overexpression of allene oxide cyclase promoted tanshinone/phenolic acid production in Salvia miltiorrhiza. Plant Cell Rep 31, 2247–2259 (2012).
Li, Y. G., Song, L., Liu, M., Hu, Z. B. & Wang, Z. T. Advancement in analysis of Salvia miltiorrhiza Radix et Rhizoma (Danshen). J Chromatogr A 1216, 1941–1953 (2009).
Gao, W. et al. Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC Genomics 15, 73 (2014).
Wang, B. et al. Genome-wide identification of phenolic acid biosynthetic genes in Salvia miltiorrhiza. Planta 241, 711–725 (2015).
Delker, C. et al. Jasmonate biosynthesis in Arabidopsis thaliana—enzymes, products, regulation. Plant Biol (Stuttg) 8, 297–306 (2006).
Koo, A. J. & Howe, G. A. The wound hormone jasmonate. Phytochemistry 70, 1571–1580 (2009).
Iván, F. A. & Edward, E. F. The Arabidopsis Book. 8 (2010).
Chini, A. et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 448, 666–671 (2007).
Thines, B. et al. JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature 448, 661–665 (2007).
Yan, Y. et al. A downstream mediator in the growth repression limb of the jasmonate pathway. Plant Cell 19, 2470–2483 (2007).
Chen, H., Jones, A. D. & Howe, G. A. Constitutive activation of the jasmonate signaling pathway enhances the production of secondary metabolites in tomato. FEBS Lett 580, 2540–2546 (2006).
Shoji, T., Ogawa, T. & Hashimoto, T. Jasmonate-induced nicotine formation in tobacco is mediated by tobacco COI1 and JAZ genes. Plant and cell physiology 49, 1003–1012 (2008).
Qi, T. et al. The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 23, 1795–1814 (2011).
Luo, H. et al. Transcriptional data mining of Salvia miltiorrhiza in response to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation. Physiol Plant 152, 241–255 (2014).
Tohge, T. et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J 42, 218–235 (2005).
Dal Cin, V. et al. Identification of genes in the phenylalanine metabolic pathway by ectopic expression of a MYB transcription factor in tomato fruit. Plant Cell 23, 2738–2753 (2011).
Gao, W. et al. Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC genomics 15, 73 (2014).
Hua, W. P., Zhang, Y., Song, J., Zhao, L. J. & Wang, Z. Z. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98, 272–279 (2011).
Yan, Y. P., Wang, Z. Z., Tian, W., Dong, Z. M. & Spencer, D. Generation and analysis of expressed sequence tags from the medicinal plant Salvia miltiorrhiza. Science China Life Sciences 53, 273–285 (2010).
Zhang, Y. et al. Pathway engineering for phenolic acid accumulations in Salvia miltiorrhiza by combinational genetic manipulation. Metabolic Engineering 21, 71–80 (2014).
Staswick, P. E. & Tiryaki, I. The oxylipin signal jasmonic acid is activated by an enzyme that conjugates it to isoleucine in Arabidopsis. Plant Cell 16, 2117–2127 (2004).
Zhang, S. et al. Cloning and Characterization of a Putative R2R3 MYB Transcriptional Repressor of the Rosmarinic Acid Biosynthetic Pathway from Salvia miltiorrhiza. PloS one 8, e73259 (2013).
Men, L., Yan, S. & Liu, G. De novo characterization of Larix gmelinii (Rupr.) Rupr. transcriptome and analysis of its gene expression induced by jasmonates. BMC Genomics 14, 548 (2013).
Yonekura-Sakakibara, K. et al. Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell 20, 2160–2176 (2008).
Vanholme, R., Demedts, B., Morreel, K., Ralph, J. & Boerjan, W. Lignin biosynthesis and structure. Plant Physiol 153, 895–905 (2010).
Wasternack, C. Jasmonates: an update on biosynthesis, signal transduction and action in plant stress response, growth and development. Annals of botany 100, 681–697 (2007).
Chung, H. S. et al. Regulation and function of Arabidopsis JASMONATE ZIM-domain genes in response to wounding and herbivory. Plant Physiol 146, 952–964 (2008).
Wager, A. & Browse, J. Social Network: JAZ Protein Interactions Expand Our Knowledge of Jasmonate Signaling. Front Plant Sci 3, 41 (2012).
Xie, D.-X., Feys, B. F., James, S., Nieto-Rostro, M. & Turner, J. G. COI1: an Arabidopsis gene required for jasmonate-regulated defense and fertility. Science 280, 1091–1094 (1998).
Stenzel, I. et al. ALLENE OXIDE CYCLASE (AOC) gene family members of Arabidopsis thaliana: tissue-and organ-specific promoter activities and in vivo heteromerization*. Journal of experimental botany 63, 6125–6138 (2012).
Sun, Y., Zhu, H., Wang, J., Liu, Z. & Bi, J. Isolation and purification of salvianolic acid A and salvianolic acid B from Salvia miltiorrhiza by high-speed counter-current chromatography and comparison of their antioxidant activity. J Chromatogr B Analyt Technol Biomed Life Sci 877, 733–737 (2009).
Ho, J. H. & Hong, C. Y. Salvianolic acids: small compounds with multiple mechanisms for cardiovascular protection. J Biomed Sci 18, 30 (2011).
Nakabayashi, R. et al. Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J 77, 367–379 (2014).
Zhang, Y., Yan, Y. P. & Wang, Z. Z. The Arabidopsis PAP1 Transcription Factor Plays an Important Role in the Enrichment of Phenolic Acids in Salvia miltiorrhiza. J Agric Food Chem 58, 12168–12175 (2010).
Etalo, D. W. et al. System-wide hypersensitive response-associated transcriptome and metabolome reprogramming in tomato. Plant Physiol 162, 1599–1617 (2013).
Pauwels, L. & Goossens, A. The JAZ proteins: a crucial interface in the jasmonate signaling cascade. Plant Cell 23, 3089–3100 (2011).
Shyu, C. et al. JAZ8 lacks a canonical degron and has an EAR motif that mediates transcriptional repression of jasmonate responses in Arabidopsis. The Plant Cell Online 24, 536–550 (2012).
Moreno, J. E. et al. Negative feedback control of jasmonate signaling by an alternative splice variant of JAZ10. Plant physiology 162, 1006–1017 (2013).
De Geyter, N., Gholami, A., Goormachtig, S. & Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends in plant science 17, 349–359 (2012).
Chini, A., Fonseca, S., Chico, J. M., Fernández‐Calvo, P. & Solano, R. The ZIM domain mediates homo‐and heteromeric interactions between Arabidopsis JAZ proteins. The Plant Journal 59, 77–87 (2009).
Oh, Y., Baldwin, I. T. & Galis, I. NaJAZh regulates a subset of defense responses against herbivores and spontaneous leaf necrosis in Nicotiana attenuata plants. Plant Physiol 159, 769–788 (2012).
Zhu, D. et al. Over-expression of a novel JAZ family gene from Glycine soja, increases salt and alkali stress tolerance. Biochem Biophys Res Commun 426, 273–279 (2012).
Feller, A., Machemer, K., Braun, E. L. & Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J 66, 94–116 (2011).
Yan, Y. P. & Wang, Z. Z. Genetic transformation of the medicinal plant Salvia miltiorrhiza by Agrobacterium tumefaciens-mediated method. Plant cell, tissue and organ culture 88, 175–184 (2007).
Fiehn, O., Kopka, J., Trethewey, R. N. & Willmitzer, L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Analytical chemistry 72, 3573–3580 (2000).
Ma, C., Wang, H., Lu, X., Xu, G. & Liu, B. Metabolic fingerprinting investigation of Artemisia annua L. in different stages of development by gas chromatography and gas chromatography-mass spectrometry. J Chromatogr A 1186, 412–419 (2008).
Yang, Y. et al. Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol Biol Rep 37, 507–513 (2010).
Doyle, J. J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry Bulletin 19, 11–15 (1987).
Zhang, Y. & Wang, Z. Z. Phenolic composition and antioxidant activities of two Phlomis species: A correlation study. C R Biol 332, 816–826 (2009).
Acknowledgements
This work benefited from financial support from the National Natural Science Foundation of China (Grant No. 31300256, Grant No. 31270338), the Natural Science Foundation of Shaanxi Province, China (Grant No. 2014JQ3100) and the “Foundation for Excellent Doctor Degree Dissertation” of Shaanxi Normal University (S2015YB02).
Author information
Authors and Affiliations
Contributions
Z.Z.W. managed and organized the study. Q.G. and Y.Z. designed the experiments and led the overall data analysis. Y.Z. and W.P.H. directed sequencing data generation and analysed the data. Y.Z. and S.H.S. carried out metabolomic analyses. Q.G., Y.C.W. and X.X.J. performed the transplants generation and functional tests. Z.Z.W., Q.G. and Y.Z. drafted the manuscript. All authors discussed and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ge, Q., Zhang, Y., Hua, WP. et al. Combination of transcriptomic and metabolomic analyses reveals a JAZ repressor in the jasmonate signaling pathway of Salvia miltiorrhiza. Sci Rep 5, 14048 (2015). https://doi.org/10.1038/srep14048
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14048
This article is cited by
-
Integrative effects of phytohormones in the phenolic acids production in Salvia verticillata L. under multi-walled carbon nanotubes and methyl jasmonate elicitation
BMC Plant Biology (2024)
-
Genome-wide analysis of the JAZ subfamily of transcription factors and functional verification of BnC08.JAZ1-1 in Brassica napus
Biotechnology for Biofuels and Bioproducts (2022)
-
Key Genes in the JAZ Signaling Pathway Are Up-Regulated Faster and More Abundantly in Caterpillar-Resistant Maize
Journal of Chemical Ecology (2022)
-
GhMYC2 activates cytochrome P450 gene CYP71BE79 to regulate gossypol biosynthesis in cotton
Planta (2022)
-
Mechanism of molecules crosstalk and the critical role of jasmonic acid on triterpenoid synthesis in Cyclocarya Paliurus cells under Aspergillus niger elicitor
Journal of Plant Biochemistry and Biotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
