
Abstract
Accumulating evidences have shown that plenty of miRNAs play fundamental and important roles in various biological processes and the deregulations of miRNAs are associated with a broad range of human diseases. However, the mechanisms underlying the dysregulations of miRNAs still have not been fully understood yet. All the previous computational approaches can only predict binary associations between diseases and miRNAs. Predicting multiple types of disease-miRNA associations can further broaden our understanding about the molecular basis of diseases in the level of miRNAs. In this study, the model of Restricted Boltzmann machine for multiple types of miRNA-disease association prediction (RBMMMDA) was developed to predict four different types of miRNA-disease associations. Based on this model, we could obtain not only new miRNA-disease associations, but also corresponding association types. To our knowledge, RBMMMDA is the first model which could computationally infer association types of miRNA-disease pairs. Leave-one-out cross validation was implemented for RBMMMDA and the AUC of 0.8606 demonstrated the reliable and effective performance of RBMMMDA. In the case studies about lung cancer, breast cancer and global prediction for all the diseases simultaneously, 50, 42 and 45 out of top 100 predicted miRNA-disease association types were confirmed by recent biological experimental literatures, respectively.
Similar content being viewed by others

Introduction
MicroRNAs (miRNAs) are a highly abundant class of small (~22 nt), endogenous and evolutionarily conserved single-stranded non-coding RNAs that regulate the gene expression normally at post-transcriptional level by base pairing with their target mRNAs at 3′ untranslated regions (UTRs), causing mRNAs degradation or translational repression1,2,3,4,5,6. As is shown in many studies, the mature miRNAs are released from a much longer RNA molecule through mainly three steps, including cropping of pri-miRNA Q1 the Drosha/DGCR8 heterodimer and producing a miRNA precursor called pre-miRNA7,8, exporting and producing a mature miRNA duplex9, dicing and degraded resulting in a fully functioning miRNA2,10,11. In 1993, lin-4, as the first miRNA, was discovered when Victor Ambros and colleagues performed a genetic screen to investigate defects in the temporal control of Caenorhabditis elegans (C. elegans) development12. Then, some years later, the second miRNA (let-7) was discovered13. After the discovery of these two well-known miRNAs, many laboratories have focused their research on these small non-coding RNAs (ncRNAs)14,15,16,17,18,19 and many miRNAs have been found in plants, green algae, viruses and animals20. According to the miRNA database (miRBase) released in 2014, there has been more than 2588 mature miRNAs in humans.
In the past few years, increasing evidences suggest that miRNAs play significant roles in a variety of essential biological processes, such as cell cycle regulation, differentiation, development, metabolism, neuronal patterning, aging and so on3. MiRNAs often play main regulator roles by binding to their target messenger RNAs (mRNAs) based on complete or partial sequence complementarity to induce transcript degradation or translational repression, respectively21. It has been demonstrated that a single miRNA can bind to multiple mRNAs and a target gene can be targeted by multiple miRNAs22. In addition to the complex base pairing between the miRNA and the mRNA, multiple miRNAs can also synergistically regulate one or more pathways23,24,25. It has been observed that miRNA-related regulations are complicated and evolutionarily conserved26,27,28. Therefore, it is no surprise that accumulating evidences show that the deregulations of miRNAs are associated with a broad range of human diseases, such as cancer, neurological disorders, cardiovascular disorders and so on29. For example, miR-17–92 cluster is shown to be oncogenic and responsible for malignant lymphoma30,31. MiR-206 can slow down the progression of ALS by sensing motor neuron injury and the miR-206 deficiency in ALS mouse model accelerate disease progression32. The miR-1 is involved in heart development and deletion of miRNA-1–2 interrupts the regulation of cardiogenesis33,34. Especially, the alterations of miRNA expression are involved in the initiation, progression and metastasis of various types of human cancers35, such as breast cancer36, lung cancer37, prostate cancer38, colon cancer39 and ovarian cancer40. Meanwhile, new disease related-miRNAs evidenced by several lines of experimental literatures are constantly emerging. Moreover, miRNAs have become a class of potential biomarkers for detection, diagnosis, prognosis and therapy in various human complex diseases41,42,43. Therefore, the disease-related miRNAs identifications are critical for not only understanding the molecular mechanisms of diverse diseases, but also providing potential biomarkers in disease diagnosis, treatment and prognosis and potential drug targets in drug discovery and clinical treatment.
Although many researchers have devoted their efforts on miRNA-disease interactions identification, the mechanisms underlying the dysregulations of miRNAs still have not been fully understood yet. Especially, the dysregulations of miRNAs cause disease through various kinds of mechanism. In the Human MicroRNA Disease Database (HMDD)44, miRNA-disease associations were divided into four types according to the different supporting evidences, including miRNA-disease association data from the evidence of genetics, epigenetics, circulating miRNAs and miRNA-target interactions, respectively. Genetic alterations (for example, SNP or deletion) and epigenetic changes (such as CpG methylation at the promoter and abnormal histone modification) may affect the transcription of the pri-miRNAs, causing aberrant expression level of the miRNAs and leading to diseases. For example, chronic lymphocytic leukemia resulted from the deletion and subsequent down-regulation of the miR-15 and miR-16 at 13q14 locus45 and methylation of the miR-200b promoter reduced its expression and was associated with metastasis or hormone receptor status in advanced breast cancer46. Nowadays, the circulating miRNAs are considered to be diagnostic biomarkers47. For example, let-7a and miR-16 were associated with the progression-free survival and overall survival of myelodysplastic syndrome (MDS) patients and could serve as noninvasive prognostic makers48. MiRNAs can bind to the UTR region of the mRNA and induce its degradation or repress its translation49. The dysregulations of the miRNA-target interactions leads to many kinds of disease, such as lung cancer induced by misregulation of miR-let-7 and its target KRAS50, Alzheimer’s disease associated with miR-103, miR-107 and their elevated target cofilin51. Furthermore, the same miRNA could be associated with the same disease based on different association types. For example, miR-137 inhibited the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. Meanwhile, miR-137 was downregulated in lung cancer cells by DNA methylation52. Therefore, lung cancer-miR-137 association was classified into both epigenetic and miRNA-target interaction types.
In the recent years, based on the assumption that miRNAs which have similar functions are often associated with similar disease and vice versa, many approaches have been proposed to predict miRNA-disease associations. For example, Jiang et al.53 developed a computational model based on hypergeometric distribution to infer potential miRNA-diseases associations by integrating the miRNA functional interactions network, disease phenotype similarity network and the known phenome-microRNAome network. Only local similarity measure has been adopted, so the prediction accuracy is not satisfactory. Shi et al.54 predict miRNA-disease associations by taking advantage of the functional links between miRNA targets and disease genes in the protein-protein interaction network. Mørk et al.21 presented a miRPD method in which not only the disease-associated miRNAs but also the underlying related proteins were predicted by combining known and predicted miRNA-protein interactions with text mined protein-disease associations. However, above three methods both strongly rely on the predicted miRNA-target interactions with a high rate of false-positive and high false-negative results. Chen et al.55 developed the first global network-based method, RWRMDA, to predict novel miRNA-disease associations by implementing random walk on the miRNA functional similarity network, which doesn’t rely on predicted miRNA-target interactions. Then, Xuan et al.56 proposed a new method named HDMP to predict potential miRNAs associated with human disease based on weighted k most similar neighbors. In that study, the previous miRNA functional similarity calculation methods were improved by incorporating the information content of disease terms, disease phenotype similarity and the information of miRNA family and cluster. RWRMDA and HDMP obtained a reliable performance for miRNA-disease association prediction, but they can’t be applied to the diseases without known related miRNAs. Furthermore, HDMP strongly rely on the selection of the number of neighbors considered in the model and it didn’t set different values of this parameter when different diseases were investigated. Based on the assumption that if miRNAs implicated in a specific tumor phenotype, their target genes will be aberrantly regulated, Xu et al.4 constructed a miRNA-target dysregulated network (MTDN) by integrating target prediction results and expression profiles data of miRNA and mRNA that in tumor and non-tumor tissues. Furthermore, feature vectors were extracted and support vector machine classifier was adopted to distinguish positive disease miRNAs from negative ones, respectively. However, this method needs the information of known negative disease-related miRNAs. By integrating the information of known miRNA-disease associations, disease-disease semantic similarity and miRNA functional similarity, Chen et al.57 further developed a semi-supervised prediction method (RLSMDA) based on the assumption that functional similar miRNAs tend to be associated with similar diseases and the framework of regularized least squares. RLSMDA achieved excellent performance in the both cross validation and case studies about several important diseases. Specially, RLSMDA is a semi-supervised method, so it does not need negative samples. More importantly, RLSMDA demonstrated excellent predictive ability for diseases without any known related miRNAs.
However, all of these previous approaches can only predict binary associations between diseases and miRNAs. On the one hand, current rich information about different types of miRNA-disease associations has not been well exploited for disease-related miRNA prediction. On the other hand, no previous computational methods could predict the types of disease-miRNA associations. Predicting the different types of disease-miRNA associations can further broaden our understanding about the molecular basis of diseases in the level of miRNAs. In this paper, we developed the model of Restricted Boltzmann machine for multiple types of miRNA-disease association prediction (RBMMMDA) to predict different types of miRNA-disease associations. Based on this model, we could obtain not only new miRNA-disease associations, but also corresponding association types. Restricted Boltzmann machine (RBM) has become the core of deep learning and successfully applied to solve various problems. Based on the following considerations, we chose RBM to predict multiple miRNA-disease associations in this work. Firstly, compared to previous work in predicting miRNA-disease associations outlined in the introduction section, RBM model could be used to predict multi-type associations and has shown its powerful performance in multi-type drug-target interaction prediction58 and neuroimaging data analysis59. Secondly, RBM provides a self-contained framework to obtain competitive classifiers directly and does not need to collect biological features and implement feature selection when classical methods such as SVM are adopted60. Thirdly, RBM could capture strong high-order non-linear correlations between the activities of features in the layer and therefore has demonstrated a good predict performance58,60,61. To our knowledge, RBMMMDA is the first model which could infer association types of miRNA-disease pairs on a large scale.
Leave-one-out cross validation (LOOCV) was implemented for RBMMMDA based on the known experimentally verified multiple types of miRNA-disease associations obtained from HMDD. As a result, the AUC of 0.8606 demonstrated the reliable and effective performance of RBMMMDA. Furthermore, RBMMMDA was evaluated by the case studies of lung cancer and breast cancer. Fifty and forty-two out of top 100 predicted miRNA-disease association types were confirmed by recent biological experimental literatures, respectively. Especially, RBMMMDA is a global approach, which can predict miRNA-disease association types for all the diseases simultaneously. Therefore, we applied RBMMMDA to all the diseases investigated in this study simultaneously and confirmed 45 out of top 100 predicted miRNA-disease association types. These confirmed associations were involved with as many as 10 important human complex diseases, such as breast cancer, hepatocellular cancer, non-small-cell lung cancer and colorectal cancer. The excellent performance in the LOOCV and case studies fully demonstrated the potential value of RBMMMDA for the identification of miRNA-disease association types and the detection of human disease biomarkers.
Results
Performance evaluation
In this paper, we implemented LOOCV on the known multiple types of miRNA-disease associations obtained from HMDD to evaluate the predictive performance of RBMMMDA. Here, we considered all the diseases simultaneously to implement the global LOOCV. As for the parameters, we chose the learning rate as 0.01 and iterative number as 100 according to previous successful study of applying the idea of Restricted Boltzmann machine (RBM) into drug-target interactions prediction58. Specifically, each known miRNA-disease association was left out in turn as test association and other known multiple types of miRNA-disease associations were taken as seed associations. After that, RBMMMDA model was trained and predictive results were provided. Then this test miRNA-disease association was ranked relative to candidate associations which include all the miRNA-disease pairs that don’t have known experimental evidences. If the rank of the test miRNA-disease association exceeds the given threshold, the RBM model was considered to predict this miRNA-disease association correctly.
Finally, Receiver-Operating Characteristics (ROC) curve which plots true positive rate (TPR, sensitivity) versus false positive rate (FPR, 1-specificity) was drawn. Sensitivity refers to the percentage of the test miRNA-disease associations which are ranked higher than the given threshold. And specificity refers to the percentage of miRNA-disease associations that are below the threshold. Then the area under ROC curve (AUC) was calculated to evaluate the performance of RBMMMDA method. If AUC = 1, it means that the RBMMMDA method has perfect performance. And AUC = 0.5 indicates random performance. As a result, RBMMMDA achieved a reliable AUC of 0.8606 (See Fig. 1). Considering RBMMMDA is the first method to predict the multiple types of miRNA-disease associations, therefore there is no other method to implement performance comparisons. However, excellent predictive ability of RBMMMDA has been demonstrated based on the above LOOCV.

Performance evaluation of RBMMMDA in term of ROC curve and AUC based on LOOCV.
As a result, RBMMMDA achieved a reliable AUC of 0.8606, demonstrating the reliable predictive ability of RBMMMDA. More importantly, RBMMMDA is the first method which could computationally predict the multiple types of miRNA-disease associations.
Case studies
Researchers in the field of computational biology and machine learning are much concerned about overfit, which means the training error would keep decreasing steadily and the generalization error would start increasing instead of decreasing. In order to see whether RBM tends to overfit, case studies have been implemented to validate the multiple types of miRNA-disease associations in the prediction list. All the known multiple types of miRNA-disease associations in the gold standard dataset were used as training samples to predict potential miRNA-disease associations and their association types for several important diseases based on the model of RBMMMDA. Prediction results were verified based on recent biological experimental results to demonstrate the prediction ability of RBMMMDA.
Breast cancer is currently regarded as the most leading type of invasive cancer in women worldwide and it is estimated that there will be approximately 231,840 new cases of invasive breast cancer and 40,290 breast cancer deaths happen among US women in 201562. The number of the affected people is still increasing, which has been predicted to reach nearly 3.2 million new cases per year by 205063. Invasive breast cancer would occur in about one eighth of the women from the United States in her lifetime. Breast cancer can also be diagnosed in men, but with a much lower ratio than that in women64. The majority deaths of the breast cancer come from the developing countries, where most of the women are diagnosed in late stages65. Recently, growing evidence shows that several miRNAs are highly correlated with breast cancer and play important roles in the tumorigenesis of breast cancer. There are 176 miRNAs known to be related to the breast cancer in the golden standard dataset and the associations of the breast cancer with related miRNA are categorized into four different subtypes according to the different supporting evidences. For example, mir-10b, which is up-regulated in metastatic breast carcinomas compared with the benign breast lesions, targets E-cadherin to promote tumor cell invasion, while mir-122 is down-regulated in breast cancer cells and functions to inhibit tumorigenesis of the cancer by targeting IGF1R66,67. This kind of miRNA and disease association is classified to be evidences from miRNA-target interaction. We implemented RBMMMDA to prioritize candidate miRNAs without the known relevance to breast cancer. As a result, among the top 10, 20 and 100 potential breast cancer-related miRNAs, 7, 13 and 42 miRNA-disease associations and their association type predications are supported by various biological experimental literatures, respectively (See Table 1 and Supplementary Table 1). It has been well-known that let-7 family mainly functions as tumor suppressors to inhibit breast cancer development and migration. Seven miRNAs from let-7 family has been ranked in top 10 predict list and five out of them has been confirmed by experimental literatures. For example, it has been confirmed that let-7i and let-7b can both inhibit the invasion of the breast cancer by targeting the oncogenes and tumor migration-related genes and induce tamoxifen sensitivity of the breast cancer by repressing the estrogen receptor α68,69,70,71,72; Down-regulation of let-7 g promotes breast cancer invasion by stimulating GAB2 and FN1 expression73; Androgen induced let-7a expression contributes to ER-, PR-, AR+ breast cancer pathogenesis74; Let-7f is a tumor-suppressor miRNA in breast cancer, which is induced by Aromatase inhibitors(Als) treatment to inhibit the aromatase gene and is also involved in low-dose metronomic (LDM) paclitaxel therapy by targeting Thrombospondin-175,76. Both mir-193b and mir-221 has been ranked in the top 10 prediction list for breast cancer and confirmed by literatures. Mir-193b decreases in breast cancer cells, which allows the expression of its target genes DNAJC13 and RAB22A and promotes breast cancer progression77. The plasma mir-221 is accumulated in breast cancer patients78 and may be a predictive biomarker for sensitivity to Neoadjuvant chemotherapy in patients with breast cancer79.
According to the American Cancer Society, the lung cancer is the most common cause of cancer deaths worldwide in both man and woman, which account for about 13% of all new cancers and 27% all cancer deaths, greater than the combination of colon, breast and prostate cancer80. There are estimated 1.4 million deaths of lung cancer each year80,81,82,83. The most affected people come from North American, Europe and East Asia. Especially, lung cancer has become the first cause of death among people with malignant tumors in China and the registered lung cancer mortality rate has increased by 464.84%84. The five-year survival rate of lung cancer is much lower than many other leading cancers, such as breast cancer and prostate cancer, due to the fact that most lung cases are diagnosed at late stage80,81,85,86,87. So it’s important and urgent to study the mechanism of the tumorigenesis of lung cancer and screen for new biomarkers for early detection80,81,88,89,90. Recently, many miRNAs have been shown to play critical roles in lung cancer development and progression91,92,93. In our golden standard dataset, there are 52 lung cancer related microRNAs with various association types. For example, mir-101 is reduced in non-small cell lung cancer (NSCLC) and can suppress NSCLC development by targeting enhancer of Zeste Homolog294, in contrast, the plasma miR-29c is significantly increased in NSCLC95. We further prioritize candidate miRNAs based on the scored calculated based on RBMMMDA. Half of top 100 potential lung cancer-related miRNAs and their association types are confirmed by several literatures. Especially, among the top 10 and top 20 prediction list, 90% of them have literature evidences (See Table 2 and Supplementary Table 2). In the top 10 potential related miRNAs, mir-34 family (a and b), which functions as tumor-suppressive miRNAs to induce apoptosis and inhibit proliferation in lung cancer cells by directly targeting TGFβR2 and Met, are inactivated by CpG methylation at their promoter region96,97,98,99,100,101,102,103; Also, mir-218, 133a and 143 are tumor-suppressors that play roles in inhibiting tumor cell invasion by targeting the tumorigenesis-related genes in lung cancer, such as N-cadherin, oncogenic receptors and so on104,105,106,107,108,109,110,111,112,113. There is also confirmed lung cancer-related miRNAs with the third association type. For example, sequence variants of mir-146a are associated with increased risk of NSCLC. Only mir-16 in the top 10 list currently has no supporting evidence.
RBMMMDA is a global ranking method, which could predict potential multiple association types of miRNA-disease pair for all the diseases simultaneously. Therefore, RBMMMDA was further applied to simultaneously rank all the candidate miRNA-disease associations. As a result, 45 of top 100 potential associations have experimental evidences (See Table 3 and Supplementary Table 3). Except for the breast and lung cancer, there are many other diseases involved in the top 100 list, such as Hepatocellular carcinoma, Stomach neoplasms, Melanoma and so on. In the top 10 prediction list, except for 7 breast cancer-related miRNAs, the regulatory mechanism of which are mentioned above, other 3 miRNAs in the top 10 list all belong to mir-34 family. MiR-34a is shown to inhibit the growth and the metastasis of gastric cancer by directly targeting Met and PDGFR114. The CpG island methylation frequency of miR-34b in hepatocellular carcinoma cancer (HCC) is significantly higher in the tumor cells compared with that in adjacent non-tumor tissues, which may correlate to the inactivation of miR-34b in HCC115. MiR-34c is predicted to be related to prostatic Neoplasms, which currently has no experimental evidence. In the top 100 association, we can further observer that one miRNA may be related to one disease based on different association types, while it may also be associated with different diseases based on the same association type. For example, miR-34a, the methylation frequency of which is significantly higher in hepatocellular carcinoma than that in the non-tumor tissues, inhibits the invasion of both stomach neoplasms and hepatocellular carcinoma by targeting Met114,115,116,117.
In conclusions, fifty, forty-two and forty-five out of top 100 predicted miRNA-disease association types for breast cancer, lung cancer and global prediction have been confirmed by recent biological experimental literatures, respectively. All of these validation results demonstrate that RBMMMDA doesn’t have a tendency of overfit.
Predicting novel multiple types of miRNA-disease associations
Here, after confirming the reliable performance of RBMMMDA in the framework of LOOCV and case studies, we further applied RBMMMDA to predict potential human miRNA-disease associations under the four different association types for all the diseases investigated in this paper. All the known multi-type miRNA-disease associations obtained from HMDD were used as training data. For all the 174 diseases, we publicly released the top 100 potential related miRNAs under four association types for each disease to facilitate experimental validation of human miRNA-disease associations. In the above case studies about breast cancer and lung cancer, most of the top 100 multiple types of miRNA-disease associations have been confirmed. Therefore it is anticipated that other potential multi-type miRNA-disease associations predicted by RBMMMDA could be validated by further biological experiments.
Discussions
Identifying novel miRNA-disease associations and their corresponding association types is vitally important goal for biological development, which plays a critical role in the understanding of disease pathogenesis at the miRNA level. In this paper, we proposed the first computational method, RBMMMDA, to predict different types of miRNA-disease associations on a large scale based on known multiple types of miRNA-disease associations derived from HMDD. RBMMMDA approach can effectively encode multiple types of miRNA-disease associations by constructing an RBM model and can effectively predict different types of miRNA-disease associations, including genetics, epigenetics, circulating miRNAs and miRNA-target interactions, respectively. The performance of RBMMMDA was evaluated by implementing LOOCV on the known experimentally verified multiple types of miRNA-disease associations. The AUC score of 0.8606 demonstrated the reliable and effective performance of RBMMMDA. Moreover, we implemented case studies of breast cancer and lung cancer for further evaluations, in which fifty and forty-two out of top 100 predicted miRNA-disease association types were confirmed by recent biological experimental literatures, respectively. More importantly, RBMMMDA was applied to predict multiple types of miRNA-disease associations for all the disease simultaneously and forty-five out of top 100 results were confirmed. All of these show the reliable performance of RBMMMDA. It is anticipated that RBMMMDA could be an important and valuable computational tool for miRNA-disease association prediction and miRNA biomarker identification for human disease diagnosis, treatment, prognosis and prevention.
The reliable performance of RBMMMDA can largely be attributed to the combination of the following several factors. Firstly, RBMMMDA takes full advantage of known multiple types of miRNA-disease associations obtained from HMDD to implement predictions, which can further help us to understanding the molecular basis of diseases in the level of miRNAs under four different association types. Secondly, as far as we know, compared with previous methods that could only predict the binary associations between miRNAs and diseases, RBMMMDA is the first computational approach for multiple types of miRNA-disease association prediction, which can not only predict potential miRNA-disease associations, but also their corresponding association types. Finally, RBMMMDA could be applied to predict miRNA-disease association types for all diseases simultaneously.
Of course, some limitations also exist in the current version of RBMMMDA. Firstly, how to choose the appropriate parameter values in RBMMMDA is not still solved well. Secondly, the current version of RBMMMDA only takes advantage of the information of known multiple types of miRNA-disease associations. In the future, new biological information, such as the disease similarity information and miRNA functional similarity, could be also incorporated into our predictive model to further improve the performance of RBMMMDA. Currently, the RBM model only considered the connections between visible layer and hidden layer, the connections within the same layers is not allowed, thus how to integrate the data of disease similarity and miRNA similarity still require careful consideration. Thirdly, RBMMMDA is not applicable to the diseases without any known miRNA-disease association information. Finally, RBMMMDA may cause bias to miRNAs with more known associated diseases. In the future, with the existence of more available experimental verified multiple types of human miRNA-disease associations, the performance of RBMMMDA will further be improved.
Methods
Multiple types of miRNA-disease associations
In this paper, we downloaded the data of miRNA-disease associations from HMDD V2.0 (http://www.cuilab.cn/hmdd) constructed by Li et al.44, which provide a comprehensive resource of experimentally verified miRNA-disease associations and lays an important data fundamental for further miRNA-related computational research. The new version of database annotates miRNA-disease associations in more details, including miRNA-disease association data from miRNA-target interactions, circulation, epigenetics and genetics. After getting rid of duplicate associations with the different evidences, we obtained 1680 distinct high-quality experimentally confirmed multi-type miRNA-disease associations about 174 diseases, 322 miRNAs and 4 different types of associations and used these miRNA-disease associations as training samples. Specifically, the data contains 682 miRNA-target interactions, 443 circulation, 199 epigenetics and 356 genetics, respectively (see Supplementary Table 4). To our knowledge, the multiple types of miRNA-disease training samples used in this study have been the largest dataset until now. Considering training samples are incomplete and no previous computational models have been developed to solve this important problem, predicting multiple types of miRNA-disease associations is a difficult challenge. However, it is worth noting that RBMMMDA still obtained the reliable predictive performance in both LOOCV (AUC of 0.8606) and case studies about lung cancer, breast cancer and global prediction for all the diseases simultaneously. More available associations obtained in the future would further improve the predictive performance of RBMMMDA.
RBMMMDA
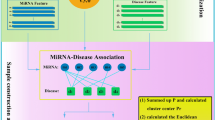
In this study, we developed the model of Restricted Boltzmann machine for multiple types of miRNA-disease association prediction (RBMMMDA) to predict different types of miRNA-disease associations (See Fig. 2, motivated by literature by Wang and Zeng58). Based on this model, we could obtain not only new miRNA-disease associations, but also their corresponding association types. RBM has been successfully applied to many important research fields58,118,119.

Flowchart of RBMMMDA, demonstrating the basic ideas of predicting multiple types of disease-miRNA association in the framework of RBM, which includes the basic there steps: constructing RBMs from a disease-miRNA interaction network; training RBM by CD algorithm; implementing prediction by computing conditional probabilities.
As shown in Fig. 2, RBM is a two-layer undirected graphical model consisting of layers of visible units and hidden units, respectively. In our RBM model, a visible unit is used to represent a disease. Hidden units represent unknown features describing miRNA-disease associations. In visible or hidden layer, there is no intra-layer connection. Furthermore, each visible unit is connected to all hidden units.
In the first step in Fig. 2, a simple example is provided to demonstrate how to construct RBMs from a multidimensional miRNA-disease interaction network. There are two miRNAs and three diseases which are included in this simple miRNA-disease network. Firstly, each miRNA-disease pair is associated with four binary variables, which indicates whether this miRNA-disease pair corresponds to this association type (i.e. the miRNA-disease associations from the evidences of miRNA-target interactions, circulation, epigenetics and genetics, respectively). Then, a particular RBM is constructed for each single miRNA. Here, two RBMs are constructed and each RBM contains three visible units representing three diseases. The binary numbers inside rectangles represent the states of visible units to indicate whether a disease and a miRNA have a connection under each specific type. RBM model captures the existed multi-type connections between disease and miRNA pair in the multidimensional miRNA-disease interaction network to implement further prediction. In this example, we have known miRNA-disease associations for miRNA 1. Therefore, three diseases send messages to hidden units and update their states and then the states of hidden units for miRNA 1 are obtained. After that, hidden units send messages again to visible units and update their states. Based on this idea, RBM is trained and potential multiple types of miRNA-disease associations are obtained.
Training RBM
In an RBM, suppose that in total there are n visible units, m hidden units and t types of miRNA-disease associations encoded in a visible unit. v = (v1, …, vn) and h = (h1, …, hm) denotes the configuration of visible layer and hidden layer, respectively. Then (v, h) is a joint configuration of an RBM. In visible layer, the binary indicator vector , denotes the state of i-th visible unit. In hidden layer, hj, j = 1, …, m denotes the state of j-th hidden unit. Let
, denotes the state of i-th visible unit. In hidden layer, hj, j = 1, …, m denotes the state of j-th hidden unit. Let  be the weight between visible variable
be the weight between visible variable  and hidden variable hj, and
and hidden variable hj, and , bj denote bias weights of visible units and hidden units, respectively. To further formulate our RBM model, a binary indicator vector r = (r1, …, rm) is adopted, in which rj = 1 if there exists a known disease-miRNA interaction between the input miRNA and the j-th disease and rj =0 otherwise. And Dij is a parameter describing the effect of r on h.
, bj denote bias weights of visible units and hidden units, respectively. To further formulate our RBM model, a binary indicator vector r = (r1, …, rm) is adopted, in which rj = 1 if there exists a known disease-miRNA interaction between the input miRNA and the j-th disease and rj =0 otherwise. And Dij is a parameter describing the effect of r on h.
Then the energy of a joint configuration (v, h) can be defined by

Then the probability of a joint configuration can be defined by

where is called the normalizing constant or partition function. Then we can get the marginal distribution over all visible data v by summing all possible configurations of h.
is called the normalizing constant or partition function. Then we can get the marginal distribution over all visible data v by summing all possible configurations of h.

According to equation (3), we can get the probability distribution over input data. In visible layer or hidden layer, there is no intra-layer connection, so we can define the following conditional probabilities:


where σ(x) = 1/(1 + e−x) is the logistic function.
However, we do not know the values of many parameters, such as  ,
,  , bj, Dij. Therefore, a mean-field version of the Contrastive Divergence (CD) algorithm is adopted here to train RBM and obtain the values of various parameters. In the CD algorithm, we use the following procedure in each training pass to incrementally adjust the weights and bias to maximize the likelihood of visible data with respect to the parameters
, bj, Dij. Therefore, a mean-field version of the Contrastive Divergence (CD) algorithm is adopted here to train RBM and obtain the values of various parameters. In the CD algorithm, we use the following procedure in each training pass to incrementally adjust the weights and bias to maximize the likelihood of visible data with respect to the parameters  ,
,  , bj and Dij.
, bj and Dij.




where  is the learning rate,
is the learning rate,  denotes an average value over all input data for each update and
denotes an average value over all input data for each update and  denotes the average value over T mean-field iteration. Based on CD algorithm, the parameters of RBM model are obtained. Therefore, we can use this RBM model to implement prediction.
denotes the average value over T mean-field iteration. Based on CD algorithm, the parameters of RBM model are obtained. Therefore, we can use this RBM model to implement prediction.
Prediction and Implementation
We can compute the following conditional probabilities after one mean-field iteration to predict the unknown interactions between disease and miRNA pair.


Because there is no intra-layer connection between any pair of visible or hidden units, once given the input data, we can get the state of hidden units according to equation (10). Then, use equation (11) we can get the probability distribution of visible units as our final prediction.
We implemented the whole algorithm in Java and used the jaRBM package. In order to implement the algorithm, we need to initialize some variables. In this study, according to previous successful study of applying RBM to potential drug-target interactions prediction58, we set the number of hidden units m = 100, learning rate ε = 0.01 and chose Gaussian distribution with standard derivation of 0.1 to initialize  , hj,
, hj,  and Dij. As for other parameters, we used the default values defined in jaRBM package.
and Dij. As for other parameters, we used the default values defined in jaRBM package.
Webserver of RBMMMDA
In addition, we built a web server which can implements the prediction function of RBMMMDA. This web server is freely available at http://42.120.43.172/RBMMMDA/. This web server enables the prediction of multiple types of miRNA-disease associations based on RBMMMDA method. When visitors choose a specific disease, potential miRNA associated with this disease based on various association types would be provided. The final prediction results would be shown in a table, where the miRNA name, association type and potential association probability would be included.
Additional Information
How to cite this article: Chen, X. et al. RBMMMDA: predicting multiple types of disease-microRNA associations. Sci. Rep. 5, 13877; doi: 10.1038/srep13877 (2015).
References
Ambros, V. The functions of animal microRNAs. Nature 431, 350–355 (2004).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism and function. Cell 116, 281–297 (2004).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Xu, J. et al. Prioritizing Candidate Disease miRNAs by Topological Features in the miRNA Target–Dysregulated Network: Case Study of Prostate Cancer. Mol Cancer Ther 10, 1857–1866 (2011).
Kong, W. et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 33, 679–689 (2014).
Li, Y., Liang, C., Wong, K.-C., Luo, J. & Zhang, Z. Mirsynergy: detecting synergistic miRNA regulatory modules by overlapping neighbourhood expansion. Bioinformatics 30, 2627–2635 (2014).
Lee, Y. et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23, 4051–4060 (2004).
Han, J. et al. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 18, 3016–3027 (2004).
Acunzo, M., Romano, G., Wernicke, D. & Croce, C. M. MicroRNA and cancer - A brief overview. Adv Biol Regul 57, 1–9 (2015).
Chendrimada, T. P. et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740–744 (2005).
Diederichs, S. & Haber, D. A. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131, 1097–1108 (2007).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993).
Reinhart, B. J. et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906 (2000).
Abrahante, J. E. et al. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell 4, 625–637 (2003).
Lagos-Quintana, M., Rauhut, R., Lendeckel, W. & Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 294, 853–858 (2001).
Lau, N. C., Lim, L. P., Weinstein, E. G. & Bartel, D. P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862 (2001).
Lee, R. C. & Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 294, 862–864 (2001).
Sempere, L. F. et al. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome. Biol 5, R13 (2004).
Vella, M. C., Choi, E.-Y., Lin, S.-Y., Reinert, K. & Slack, F. J. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′ UTR. Genes Dev 18, 132–137 (2004).
Griffiths-Jones, S., Saini, H. K., Van Dongen, S. & Enright, A. J. miRBase: tools for microRNA genomics. Nucleic Acids Res 36, D154–D158 (2008).
Mørk, S., Pletscher-Frankild, S., Caro, A. P., Gorodkin, J. & Jensen, L. J. Protein-driven inference of miRNA–disease associations. Bioinformatics 30, 392–397 (2014).
Yang, H. et al. Evaluation of genetic variants in microRNA-related genes and risk of bladder cancer. Cancer Res 68, 2530–2537 (2008).
Boross, G., Orosz, K. & Farkas, I. J. Human microRNAs co-silence in well-separated groups and have different predicted essentialities. Bioinformatics 25, 1063–1069 (2009).
Krek, A. et al. Combinatorial microRNA target predictions. Nat Genet 37, 495–500 (2005).
Xu, J. et al. MiRNA–miRNA synergistic network: construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res 39, 825–836 (2011).
Chen, K. & Rajewsky, N. Deep conservation of microRNA-target relationships and 3′UTR motifs in vertebrates, flies and nematodes. Cold Spring Harb Symp Quant Biol 71, 149–156 (2006).
Li, J. et al. Evidence for positive selection on a number of microRNA regulatory interactions during recent human evolution. PLoS Genet 8, e1002578 (2012).
Xie, X. et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434, 338–345 (2005).
Hatziapostolou, M. et al. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147, 1233–1247 (2011).
He, L. et al. A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 (2005).
Tagawa, H. & Seto, M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia 19, 2013–2016 (2005).
Williams, A. H. et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326, 1549–1554 (2009).
Yang, B. et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13, 486–491 (2007).
Zhao, Y. et al. Dysregulation of cardiogenesis, cardiac conduction and cell cycle in mice lacking miRNA-1-2. Cell 129, 303–317 (2007).
Volinia, S. et al. Reprogramming of miRNA networks in cancer and leukemia. Genome Res 20, 589–599 (2010).
Iorio, M. V. et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65, 7065–7070 (2005).
Yanaihara, N. et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189–198 (2006).
Porkka, K. P. et al. MicroRNA expression profiling in prostate cancer. Cancer Res 67, 6130–6135 (2007).
Akao, Y., Nakagawa, Y. & Naoe, T. MicroRNA-143 and-145 in colon cancer. DNA Cell Biol. 26, 311–320 (2007).
Yang, H. et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 68, 425–433 (2008).
Cho, W. C. MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int. J. Biochem. Cell Biol 42, 1273–1281 (2010).
Link, A. et al. Fecal MicroRNAs as novel biomarkers for colon cancer screening. Cancer Epidemiol Biomarkers Prev 19, 1766–1774 (2010).
Tricoli, J. V. & Jacobson, J. W. MicroRNA: potential for cancer detection, diagnosis and prognosis. Cancer Res 67, 4553–4555 (2007).
Li, Y. et al. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res 42, D1070–D1074 (2014).
Calin, G. A. et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99, 15524–15529 (2002).
Wee, E. et al. Mapping the regulatory sequences controlling 93 breast cancer-associated miRNA genes leads to the identification of two functional promoters of the Hsa-mir-200b cluster, methylation of which is associated with metastasis or hormone receptor status in advanced breast cancer. Oncogene 31, 4182–4195 (2012).
Schwarzenbach, H., Nishida, N., Calin, G. A. & Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol 11, 145–156 (2014).
Zuo, Z. et al. Circulating microRNAs let-7a and miR-16 predict progression-free survival and overall survival in patients with myelodysplastic syndrome. Blood 118, 413–415 (2011).
Esteller, M. Non-coding RNAs in human disease. Nat Rev Genet 12, 861–874 (2011).
Chin, L. J. et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non–small cell lung cancer risk. Cancer Res 68, 8535–8540 (2008).
Yao, J. et al. MicroRNA-related cofilin abnormality in Alzheimer’s disease. PLoS One 5, e15546 (2010).
Zhu, X. et al. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett 587, 73–81 (2013).
Jiang, Q. et al. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst Biol 4, S2 (2010).
Shi, H. et al. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst Biol 7, 101 (2013).
Chen, X., Liu, M.-X. & Yan, G.-Y. RWRMDA: predicting novel human microRNA–disease associations. Mol Biosyst 8, 2792–2798 (2012).
Xuan, P. et al. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS One 8, e70204 (2013).
Chen, X. & Yan, G.-Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci Rep 4, 5501 (2014).
Wang, Y. & Zeng, J. Predicting drug-target interactions using restricted Boltzmann machines. Bioinformatics 29, i126–i134 (2013).
Hjelm, R. D. et al. Restricted Boltzmann machines for neuroimaging: an application in identifying intrinsic networks. NeuroImage 96, 245–260 (2014).
Larochelle, H. & Bengio, Y. Classification using discriminative restricted Boltzmann machines. The 25th International Conference on Machine Learning, Helsinki. New York: ACM (2008-07-05)
Salakhutdinov, R., Mnih, A. & Hinton, G. Restricted Boltzmann machines for collaborative filtering. The 24th Annual International Conference on Machine Learning, Oregon. New York: ACM (2007-06-20)
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2015. CA Cancer J Clin 65, 5–29 (2015).
Tao, Z. et al. Breast Cancer: Epidemiology and Etiology. Cell Biochem Biophys (2014).
Thorlacius, S. et al. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nature genetics 13, 117–119 (1996).
Kelsey, J. L. & Horn-Ross, P. L. Breast cancer: magnitude of the problem and descriptive epidemiology. Epidemiologic reviews 15, 7–16 (1992).
Liu, Y. et al. MicroRNA-10b targets E-cadherin and modulates breast cancer metastasis. Med Sci Monit 18, BR299–BR308 (2012).
Wang, B., Wang, H. & Yang, Z. MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS One 7, e47053 (2012).
Chen, F. et al. Let-7b inhibits human cancer phenotype by targeting cytochrome P450 epoxygenase 2J2. PloS One 7, e39197 (2012).
Hu, X. et al. The heterochronic microRNA let-7 inhibits cell motility by regulating the genes in the actin cytoskeleton pathway in breast cancer. Mol Cancer Res 11, 240–250 (2013).
Ma, L., Li, G.-z., Wu, Z.-s. & Meng, G. Prognostic significance of let-7b expression in breast cancer and correlation to its target gene of BSG expression. Med Oncol 31, 773 (2014).
Subramanian, M. et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 34, 1094–1104 (2015).
Zhao, Y. et al. let-7 microRNAs induce tamoxifen sensitivity by downregulation of estrogen receptor α signaling in breast cancer. Mol Med 17, 1233–1241 (2011).
Qian, P. et al. Pivotal role of reduced let-7g expression in breast cancer invasion and metastasis. Cancer Res 71, 6463–6474 (2011).
Lyu, S. et al. Androgen receptor decreases CMYC and KRAS expression by upregulating let-7a expression in ER-, PR-, AR+ breast cancer. Int J Oncol 44, 229–237 (2014).
Shibahara, Y. et al. Aromatase inhibitor treatment of breast cancer cells increases the expression of let-7f, a microRNA targeting CYP19A1. J Pathol 227, 357–366 (2012).
Tao, W.-Y., Liang, X.-S., Liu, Y., Wang, C.-Y. & Pang, D. Decrease of Let-7f in Low-Dose Metronomic Paclitaxel Chemotherapy Contributed to Upregulation of Thrombospondin-1 in Breast Cancer. Int J Biol Sci 11, 48–58 (2015).
Yang, Z. et al. Tumor suppressive microRNA-193b promotes breast cancer progression via targeting DNAJC13 and RAB22A. Int J Clin Exp Pathol 7, 7563–7570 (2014).
Gezer, U. et al. Abundant circulating microRNAs in breast cancer patients fluctuate considerably during neoadjuvant chemotherapy. Oncol Lett 8, 845–848 (2014).
Zhao, R. et al. Plasma miR-221 as a predictive biomarker for chemoresistance in breast cancer patients who previously received neoadjuvant chemotherapy. Onkologie 34, 675–680 (2011).
Xue, Z., Wen, J., Chu, X. & Xue, X. A microRNA gene signature for identification of lung cancer. Surg Oncol 23, 126–131 (2014).
Wang, J., Zhao, Y., Lu, Y. & Ma, C. Integrated bioinformatics analyses identify dysregulated miRNAs in lung cancer. Eur Rev Med Pharmacol Sci 18, 2270–2274 (2014).
Jemal, A., Siegel, R., Xu, J. & Ward, E. Cancer statistics, 2010. CA Cancer J Clin 60, 277–300 (2010).
Brambilla, E., Travis, W. D., Colby, T., Corrin, B. & Shimosato, Y. The new World Health Organization classification of lung tumours. Eur Respir J 18, 1059–1068 (2001).
She, J., Yang, P., Hong, Q. & Bai, C. Lung cancer in China: challenges and interventions. CHEST Journal 143, 1117–1126 (2013).
Scott, W. J., Howington, J., Feigenberg, S., Movsas, B. & Pisters, K. Treatment of non-small cell lung cancer stage I and stage II: ACCP evidence-based clinical practice guidelines. Chest 132, 234S–242S (2007).
van Zandwijk, N. Neoadjuvant strategies for non-small cell lung cancer. Lung Cancer 34, S145–S150 (2001).
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2013. CA Cancer J Clin 63, 11–30 (2013).
Swensen, S. J. CT screening for lung cancer. AJR Am J Roentgenol 179, 833–836 (2002).
Cagle, P. T. & Allen, T. C. Lung cancer genotype-based therapy and predictive biomarkers: present and future. Arch Pathol Lab Med 136, 1482–1491 (2012).
Lam, W. K. & Watkins, D. N. Lung cancer: future directions. Respirology 12, 471–477 (2007).
Yu, H. et al. Decreased circulating miR-375: a potential biomarker for patients with non-small-cell lung cancer. Gene 534, 60–65 (2014).
Wan, L., Zhang, L., Fan, K. & Wang, J. MiR-27b targets LIMK1 to inhibit growth and invasion of NSCLC cells. Mol Cell Biochem 390, 85–91 (2014).
Xiong, S. et al. MicroRNA-7 inhibits the growth of human non-small cell lung cancer A549 cells through targeting BCL-2. Int J Biol Sci 7, 805–814 (2011).
Zhang, J.-g., Guo, J.-F., Liu, D.-L., Liu, Q. & Wang, J.-J. MicroRNA-101 exerts tumor-suppressive functions in non-small cell lung cancer through directly targeting enhancer of zeste homolog 2. J Thorac Oncol 6, 671–678 (2011).
Heegaard, N. H. et al. Circulating micro‐RNA expression profiles in early stage nonsmall cell lung cancer. Int J Cancer 130, 1378–1386 (2012).
Wang, Z. et al. DNA hypermethylation of microRNA-34b/c has prognostic value for stage I non-small cell lung cancer. Cancer Biol Ther 11, 490–496 (2011).
Lodygin, D. et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7, 2591–2600 (2008).
Nadal, E. et al. Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin Cancer Res 19, 6842–6852 (2013).
Tanaka, N. et al. Frequent methylation and oncogenic role of microRNA-34b/c in small-cell lung cancer. Lung Cancer 76, 32–38 (2012).
Ma, Z.-L. et al. MicroRNA-34a inhibits the proliferation and promotes the apoptosis of non-small cell lung cancer H1299 cell line by targeting TGFβR2. Tumour Biol 36, 2481–2490 (2015).
Yu, G., Zhong, N., Chen, G., Huang, B. & Wu, S. Downregulation of PEBP4, a target of miR-34a, sensitizes drug-resistant lung cancer cells. Tumour Biol 35, 10341–10349 (2014).
Zhou, J.-Y. et al. MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting MET. Cancer Lett 351, 265–271 (2014).
Wang, L.-G. et al. MicroRNA-34b functions as a tumor suppressor and acts as a nodal point in the feedback loop with Met. Int J Oncol 42, 957–962 (2013).
Wu, D.-W., Cheng, Y.-W., Wang, J., Chen, C.-Y. & Lee, H. Paxillin predicts survival and relapse in non–small cell lung cancer by microRNA-218 targeting. Cancer Res 70, 10392–10401 (2010).
Zhang, C., Ge, S., Hu, C., Yang, N. & Zhang, J. MiRNA-218, a new regulator of HMGB1, suppresses cell migration and invasion in non-small cell lung cancer. Acta Biochim Biophys Sin (Shanghai) 45, 1055–1061 (2013).
Sher, Y.-P. et al. ADAM9 up-regulates N-cadherin via miR-218 suppression in lung adenocarcinoma cells. PloS One 9, e94065 (2014).
Tan, W., Gu, J., Huang, M., Wu, X. & Hildebrandt, M. A. Epigenetic analysis of microRNA genes in tumors from surgically resected lung cancer patients and association with survival. Mol Carcinog (2014).
Mataki, H. et al. Downregulation of the microRNA-1/133a cluster enhances cancer cell migration and invasion in lung-squamous cell carcinoma via regulation of Coronin1C. J Hum Genet 60, 53–61 (2015).
Wang, L.-K. et al. MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PloS One 9, e96765 (2014).
Moriya, Y. et al. Tumor suppressive microRNA-133a regulates novel molecular networks in lung squamous cell carcinoma. J Hum Genet 57, 38–45 (2012).
Wei, J. et al. miR?143 inhibits cell proliferation by targeting autophagy?related 2B in non?small cell lung cancer H1299 cells. Mol Med Rep 11, 571–576 (2015).
Xia, H. et al. miR-143 Inhibits NSCLC Cell Growth and Metastasis by Targeting Limk1. Int J Mol Sci 15, 11973–11983 (2014).
Zhang, N., Su, Y. & Xu, L. Targeting PKCε by miR-143 regulates cell apoptosis in lung cancer. FEBS Lett 587, 3661–3667 (2013).
Peng, Y., Guo, J.-J., Liu, Y.-M. & Wu, X.-L. MicroRNA-34A inhibits the growth, invasion and metastasis of gastric cancer by targeting PDGFR and MET expression. Biosci Rep 34 (2014).
Xie, K. et al. Methylation-associated silencing of microRNA-34b in hepatocellular carcinoma cancer. Gene 543, 101–107 (2014).
Dang, Y., Luo, D., Rong, M. & Chen, G. Underexpression of miR-34a in hepatocellular carcinoma and its contribution towards enhancement of proliferating inhibitory effects of agents targeting c-MET. PloS One 8, e61054 (2013).
Li, N. et al. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett 275, 44–53 (2009).
Hinton, G. E. & Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science 313, 504–507 (2006).
Eickholt, J. & Cheng, J. Predicting protein residue–residue contacts using deep networks and boosting. Bioinformatics 28, 3066–3072 (2012).
Acknowledgements
The financial support from the National Natural Science of Foundation of China under Grant No. 11301517, 61472203, 61327902 and National Center for Mathematics and Interdisciplinary Sciences, CAS and State Key Laboratory of Intelligent Control and Decision of Complex Systems, Beijing Institute of Technology is highly appreciated.
Author information
Authors and Affiliations
Contributions
X.C. conceived the project, developed the prediction method, designed and implemented the experiments, analyzed the result and wrote the paper. C.G.Y. implemented the experiments and analyzed the result. X.T.Z. implemented the experiments, analyzed the result and wrote the paper. ZH.L. analyzed the result and wrote the paper. L.X.D. implemented the experiments. Y.D.Z. and Q.H.D. analyzed the result. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, X., Clarence Yan, C., Zhang, X. et al. RBMMMDA: predicting multiple types of disease-microRNA associations. Sci Rep 5, 13877 (2015). https://doi.org/10.1038/srep13877
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13877
This article is cited by
-
Predicting miRNA-disease associations based on graph attention network with multi-source information
BMC Bioinformatics (2022)
-
DCTGM: A Novel Dual-channel Transformer Graph Model for miRNA-disease Association Prediction
Cognitive Computation (2022)
-
DBNLDA: Deep Belief Network based representation learning for lncRNA-disease association prediction
Applied Intelligence (2022)
-
MiRNA-disease association prediction via hypergraph learning based on high-dimensionality features
BMC Medical Informatics and Decision Making (2021)
-
RSCMDA: Prediction of Potential miRNA–Disease Associations Based on a Robust Similarity Constraint Learning Method
Interdisciplinary Sciences: Computational Life Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
