
Abstract
Filamentous bacteria are a normal and necessary component of the activated sludge wastewater treatment process, but the overgrowth of filamentous bacteria results in foaming and bulking associated disruptions. Bacteriophages, or phages, were investigated for their potential to reduce the titer of foaming bacteria in a mixed-microbial activated sludge matrix. Foaming-associated filamentous bacteria were isolated from activated sludge of a commercial wastewater treatment plan and identified as Gordonia species by 16S rDNA sequencing. Four representative phages were isolated that target G. malaquae and two un-named Gordonia species isolates. Electron microscopy revealed the phages to be siphophages with long tails. Three of the phages - GordTnk2, Gmala1 and GordDuk1 - had very similar ~76 kb genomes, with >93% DNA identity. These genomes shared limited synteny with Rhodococcus equi phage ReqiDocB7 and Gordonia phage GTE7. In contrast, the genome of phage Gsput1 was smaller (43 kb) and was not similar enough to any known phage to be placed within an established phage type. Application of these four phages at MOIs of 5–15 significantly reduced Gordonia host levels in a wastewater sludge model by approximately 10-fold as compared to non-phage treated reactors. Phage control was observed for nine days after treatment.
Similar content being viewed by others


Introduction
The activated sludge process involves the biological conversion of solids and pollutants and is an established method for sewage and industrial wastewater treatment worldwide1,2. Activated sludge, the essential component of this process, includes a mixed and variable consortium of micro- and macro-organisms including viruses, bacteria, protozoa and metazoa who function in the transformation of organic materials into a liquid mixture that is relatively low in suspended solids and organic compounds1,3. Among the key organic degraders, filamentous bacteria bond to other floc-forming organisms with biopolymers and provide important floc “backbones” to support the structure and shape of compact flocs for efficient sludge settling2. The excessive growth of filamentous bacteria, however, trap and stabilize air bubbles when combined with biosurfactants, resulting in stable sludge foaming and biomass bulking on the surface of aerated reactors. Sludge foaming is a frequent operational problem and prevents adequate flocculation and impedes proper sludge mass settling1,4. Filamentous bacteria associated with activated sludge and foaming have been extensively characterized based on filament morphotypes5,6,7, fluorescence in situ hybridization using rRNA-targeted probes8,9,10,11 and high-throughput deep-sequencing-based population analysis12,13,14,15. Accumulated studies revealed the diverse compositions of filamentous bacteria involved in foaming and suggested the common roles of long unbranched filaments of Candidatus ‘Microthrix parvicella’ or short branched filaments of mycolic acid-containing actinomycetes, or Mycolata3,16. Mycolata associated with sludge foaming include the genera Corynebacterium, Dietzia, Gordonia, Mycobacterium, Nocardia, Rhodococcus, Skermania, Tsukamurella and Williamsia17. Gordonia is frequently reported to be the predominant genus within the foaming-associated mycolata18,19, although the high abundance of Gordonia is not the sole determinant of a foaming event15.
Current control methods for sludge foaming reply on physio-chemical methods, including return sludge flow rate and aeration manipulation, surfactants and chlorine addition. These efforts are not always effective and run the risk of secondary perturbations to the sustainable sludge process20. An alternative treatment approach would be the use of bacteriophage, is capable of specifically targeting the foam causing organisms without causing collateral damage to other microorganisms21,22. Phages are a normal component of the wastewater environment23,24. The idea of using phages as antimicrobial agents, dating back to the earliest days of phage research, is an area of recently renewed interest25,26. Most of the recent research in phage-based antimicrobials has focused on pathogens of medical or agricultural importance27,28. The use of phages for the control of problem bacteria in environmental and/or industrial settings also has great potential in replacing inefficient and often environmentally harmful biocides.
Phages against filamentous bacteria, especially different species of mycolata, have been isolated and their therapeutic applications documented in several studies. Mycobacteria species in particular have been the subject of focused efforts29,30. Multiple phages against Rhodococcus equi were characterized and demonstrated to be capable of reducing R. equi load in a soil matrix31. Phages isolated from a wastewater treatment plant were shown to have promising potential in controlling biomass bulking caused by the filamentous bacteria Haliscomenobacter hydrossis32. Sludge – associated Mycolata hosts for which phages have been identified include Tsukamurella spp., Gordonia, Rhodococcus, Nocardia22,33,34,35. Two of these phages - GTE2 and GTE7 - were demonstrated to reduce their host level and prevent stable foam production by their host bacteria in pure laboratory cultures tested in a laboratory foaming apparatus33,36. Beyond these promising results, the next step in the development of phage-based foam control method is to determine if phage can control foaming associated bacteria directly in the complex chemical and microbial environment of the activated sludge matrix. Additionally, the value of a phage for bacterial control needs to be determined in reference to how broad each phage host range is against wastewater treatment plant-associated hosts. Here, filamentous bacteria were cultured from several wastewater treatment plant foaming outbreaks and used to isolate cognate phages capable of controlling these bacteria. The isolated phages were evaluated for use in reducing Gordonia levels in raw activated sludge.
Results
Isolation of Gordonia from foaming activated sludge
Activated sludge foam samples were collected from aeration basins from a local wastewater treatment plant. Phase contrast microscopic examination revealed that the samples contained compact flocs of intermeshing, true branching filamentous organisms (Supplementary Fig. S1), a typical morphological feature associated with nocardioform bacteria1. The foam samples were plated onto both semi-selective and non-selective agar plates for filamentous bacteria isolation and purification. Seven isolates exhibiting filamentous cell growth morphologies (formed short hyphae) during colony development were collected (Supplementary Fig. S1). All isolates developed colonies with irregular margins and appeared white to beige at the beginning of incubation. Strain G1 produced a pink pigment and G5 and G11 produced yellow and orange/red pigment, respectively, over prolonged incubation. Analysis of the 16s rDNA sequences (~1200 bp) of these isolates revealed four clusters, suggesting four taxonomical groups. By comparing to bacterial type strains in the database of the Ribosomal Database Project (rdp.cme.msu.edu), a phylogenetic tree was generated (Fig. 1). All seven isolates were identified at the genus level as Gordonia. Identification to the species level was suggested in the case of isolates G4 (G. malaquae), G8 and G10 (G. amarae). The 16S sequences of G5 and G11 were highly similar (>99.5%) to G. sputi, G. aichiensis and G. otitidis, and so could only be confidently identified to the genus level. The remaining two isolates, G1 and G7, were also identified only to the genus level due to their closest match (G. araii) being below the desired 99.5% cutoff. The sequenced regions of isolates G1 and G7 were identical to each other and most similar to G. araii (Fig. 1). Species designation solely based on 16sDNA sequences is limited and its accuracy needs to be confirmed by further phenotypical characterization.

Phylogenetic tree derived from 16S rDNA gene sequences, created using the neighbour-joining method with gap positions excluded.
Numbers on the tree indicate bootstrap percentages (from 1000 replicates) for branch points. Assignments of identities of the Gordonia isolates are indicated. Strains G5 and G11 were highly similar to G. sputi, G. aichiensis and G. otitidis and thus were identified to the genus level only.
Isolation and characterization of Gordonia phages
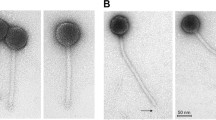
Fourteen phages were isolated from wastewater using a panel of the seven Gordonia strains described above for phage enrichments. The host range of each phage was tested at its routine test dilution and phages were found to form plaques on 4 of the 7 strains (Table 1). Based on host range, the seven phages could be grouped into four groups, of which only groups 1 and 2 were capable of attacking more than one of the strains (Table 1). Of the strains in the panel, Gordonia sp. strain G7 was the most susceptible, being sensitive to phages of groups 1–3. The phages within each host range group generated identical patterns based on restriction fragment length polymorphism (RFLP) using the enzymes EcoRI, BamHI and HindIII (data not shown). GordTnk2, Gmala1, GordDuk1 and Gsput1, were picked to represent the four phage groups for further testing. The morphologies of the four phages were determined by TEM (Fig. 2). All four phages possessed icosahedral capsids and long, non-contractile tails of siphophages. Three phages, GordTnk2, GMala1, GordDuk1 shared similar average head diameters (72–75 nm) and extremely long tail lengths (~520 nm). Phage Gsput1 was significantly smaller, with head diameter and tail length of 62 ± 1 nm and 229 ± 5 nm, respectively (Fig. 2 and Table 2).

Transmission electron micrographs of phages (A) GordTnk2, (B) Gmala1, (C) GordDuk1 and (D) Gsput1.
Samples were stained with 2% (w/v) uranyl acetate and observed at 100 kV. Scale bar is 100 nm.
Genomes of GordTnk2, Gmala1 and GordDuk1
The genomic DNA of the four Gordonia phages were sequenced (Supplementary Tables S1, S2, S3 and S4). Consistent with their similar morphologies, phages GordTnk2, Gmala1 and GordDuk1 were quite similar in genome size (75 ~ 76 kb, encoding 90–98 proteins), with high overall DNA identity (93%–95%) (Table 2). All three phage genomic termini were determined to be pac-type with limited circular permutation, based on a combination of restriction mapping and analysis of the shotgun assembly, which resolved into a single circularly permuted contig (data not shown). The core genome of phages GordTnk2, Gmala1 and GordDuk1 consisted of 90 protein-coding genes present in all three genomes. Gene-content differences between the three phages included 9 variant genes located in two regions of small, primarily hypothetical novel proteins in the left arm (data not shown). Notable similarities were found between the genomes and morphologies of these three phages and two previously described phages, Rhodococcus equi phage ReqiDocB7 (GenBank NC_023706) and Gordonia phage GTE7 (GenBank NC_016166) (Figs 2 and 3)31,33. The similarity between the three new Gordonia phages, ReqiDocB7 and GTE7 was based on overall morphological characteristics, most notably the long, flexible tails with lengths of 438–520 nm and genome arrangement, rather than nucleotide sequence. Out of 98 unique proteins, phages GordTnk2, Gmala1 and GordDuk1 shared 28 with DocB7 and 26 with GTE7, albeit all at less than 51% identity (Fig. 3A). One of the more unusual features present in all five phage genomes is a long region upstream of the right terminus with multiple inverted repeats and no protein coding genes. This region is ~1,800 bp in GordTnk2, Gmala1 and GordDuk1, which is intermediate in length to the analogous region of GTE7 (1,185 bp) and ReqiDocB7 (2,484 bp). Such large stretches of non-coding sequence are unusual in phage genomes. Another shared feature of these phages is the complexity of the predicted lysis genes. Between genes 22 and 33 of GordTnk2, Gmala1 and GordDuk1, there are four genes (22, 24, 26 and 33) whose products are predicted to function in cell wall hydrolysis and three genes encoding proteins with transmembrane domains (TMDs) (gp27, gp30 and gp31) that could function as membrane-permeabilizing holins (Fig. 4 and Table 3).

Genome map of Gordonia phages GordTnk2 and Gsput1.
GordTnk2 is presented to represent Gmala1 and GordDuk1, as these three phages share great similarity in genome arrangement. Predicted genes are represented by boxes above and below the heavy black line; boxes above the lines are genes encoded on the forward strand and those below the lines are on the reverse strand. The ruler below the genomes indicates the scale (in kb). Genome features are color coded according to the legend. (A) Comparison map of GordTnk2 to Gordonia phage GTE7 (accession No. NC_016166) and Rhodococcus equi phage ReqiDocB7 (accession No. NC_023706). Proteins sharing identities (e value < 10−5) among three phages were identified and color coded according to the legend. (B) Genome map of Gsput1. Proteins sharing identities (e value < 10−5) with Mycobacterium phage were identified and the clusters or singletons (Sgn) of the Mycobacterium phage are indicated in red letters. Abbreviations: TerS, terminase small subunit; TerL, terminase large subunit; tmp, tape measure protein; Prim, primase; Pol, DNA polymerase; DNA MTase, DNA methylase; HNH, homing endonuclease; Int, integrase; vWA, Von Willebrand factor type A domain protein; AAA, ATP-hydrolyzing domain protein; RecA, recombinase; Nuc, nuclease.

Lysis genes in Gordonia phages GordTnk2, Gmala1 and GordDuk1 and their comparisons to Gordonia phage GTE7 and Rhodococcus equi phage ReqiDocB7.
Predicted genes are represented by boxes above and below the black line (encoded on the forward and reverse strand, respectively). The ruler below the genes indicates the scale (in kb). Genome features are color coded according to the legend. Lysis and holin proteins sharing identities (e value < 10−5) among presented phages were linked with blue lines. The number of predicted transmembrane domains in holin is indicated above the protein.
For the purposes of bacterial control, virulent phages (i.e., phages unable to form lysogens) are preferred37. Based on analysis of the genomes of phages GordTnk2, Gmala1 and GordDuk1, no evidence was found suggesting a temperate lifestyle in these phages. These phages were not found to be closely related to known temperate phages or to any prophage elements incorporated into sequenced bacterial genomes. A tyrosine recombinase was identified (gp72 in GordTnk2) and such recombinases can function as phage integrases. However the small size of gp72 and its relationship to bacterial XerC-like recombinases suggests this protein is more likely to function as a general recombinase during phage DNA replication. Efforts to isolate lysogens of phages GordTnk2, Gmala1 and GordDuk1 were unsuccessful (data not shown). Taken together, the data support a model in which phages GordTnk2, Gmala1 and GordDuk1 are virulent phage not capable of lysogeny.
Genome of Gsput1
The 43,505 bp genome of Gsput1 was also determined and predicted to encode 71 proteins (Table 2, Fig. 3B). Gsput1 was found to be unrelated to phages GordTnk2, Gmala1 and GordDuk1 at both the DNA and protein levels. The Gsput1 genomic DNA was determined to possess 16 bp 5′ extended cos termini with the sequence 5′-GCTGACCCACGGACAC-3′. There was no DNA-level similarity between Gsput1 and any phage in the public database. Of 71 proteins, 41 proteins were found to share sequence similarities to previously identified proteins, primarily those encoded by mycobacteriophage. However, there were no extensive similarities to any single organism; the most closely related phage genome was that of mycophage Redi (NC_023730, eight similar proteins) and the most closely related prophage elements are located in Nocardia farcinica (NC_006361, 15 similar proteins across three genomic loci) and Corynebacterium diptheriae (BX248353, 12 proteins across three loci). To date, some 891 mycophage genomes have been determined and organized into 21 clusters and nine singletons based on nucleotide similarity (http://phagesdb.org/clusters/)30,38,39. Individual Gsput1-encoded proteins were similar to proteins distributed among 27 of the 30 mycobacteriophage clusters/singletons (Fig. 3B). Gsput1 is probably temperate, as it encodes a predicted integrase (gp28) which shares strong homology with integrase proteins found in the genomes of other Mycolata (E < 10−100) and with prophage integrases (e.g., Lactobacillus prophage Lj965 E = 10−17). Additionally, Gpsut1 gp29 encoded directly upstream of gp28, contains a detectable excisionase (Xis) domain (TIGR01764), further supporting the prediction of a functional Int/Xis module and temperate lifestyle.
Phage efficacy test in a model activated sludge system
Preliminary standard phage host killing assays in defined liquid media demonstrated that treatment of pure Gordonia cultures with phages controlled host cell growth (data not shown). The capacity of the four phages to affect Gordonia accumulation in the presence of the mixed chemistry and microbiology of activated sludge was examined. Activated sludge was collected from the local wastewater treatment plant and split into separate aerated bioreactors to start the experiment immediately. Parallel bioreactors for each treatment (control or phage treated) were set up in order to conduct the experiment in duplicate. The sludge was inoculated with Gordonia hosts and phage treatments were added two hours after the host inoculation. Dynamics of total bacteria and Gordonia accumulation, expressed as a function of genome equivalents (GEq) per ml sludge, as well as the relative abundance of Gordonia as compared to total bacteria in the bioreactors, was monitored over a nine-day period by RT-qPCR. The level of total bacteria in the fresh sludge, prior to inoculation, was measured at 5.8 × 107 GEq/ml sludge. Gordonia host background levels in the sludge were measured and native Gordonia sp. G11-like organisms were not detected while Gordonia sp. G7-like organisms were determined to be present at 8 × 104 GEq/ml sludge (data not shown). In both control and phage-treated systems after the start of the experiment, the total bacteria level increased by ~25 fold during day one, rising from 5.9 × 107 to ~2 × 109 GEq/ml sludge. Following this initial increase, total bacteria levels decreased between days one to three and reached a level around 2 × 108 GEq/ml sludge by day three and onwards (Fig. 5A). The inoculated Gordonia host strains exhibited different dynamics than did total bacteria. Following inoculation into the mixed sludge bioreactors, the host Gordonia strains exhibited an initial fluctuation in levels, primarily during the first day. By day two, both hosts stabilized and thus allowed observation of phage effect on Gordonia levels over the nine-day course of the experiment (Fig. 5B–E). In the phage treated systems, both G7 and G11 levels remained essentially constant after day two, however significant growth of G7 and G11 with different dynamics was observed in the non-phage treated systems. Gordonia G11 levels at the end of nine days were on average 1.6 × 107 GEq/ml sludge, or 12.0% of total bacteria. This is over 10-fold higher than G11 levels in phage-treated reactors, at 4.0 × 106 GEq/ml sludge, or 0.7% of total bacteria (Fig. 5B,D). In contrast, host G7 did not grow as well in the bioreactors as did G11, with the highest cell levels observed at day seven, which was followed by a steady decrease. Because of the reduced growth of G7, even though phage-treated samples showed little increase in host G7 level, on both GEq/ml and percentage of total population basis, the difference between phage treatment and control was less pronounced. Despite this, the G7 level in untreated sludge at day nine of the experiment (7.8 × 105 GEq/ml sludge, or 0.6% of total bacteria) was still higher than the G7 level in the phage treated sludge (<2.0 × 105 GEq/ml sludge, or <0.1% of total bacteria) (Fig. 5C,E).

Effect of phage on total bacterial and Gordonia levels in laboratory-scale activated sludge systems.
Gordonia strains G11 and G7 were inoculated to activated sludge with (empty mark) or without (filled mark) phage cocktail treatment. Quantities (expressed as genome equivalents, or GEq per ml sludge) of total bacterial (A), Gordonia G11 (B), Gordonia G7 (C), percentage of total bacteria of Gordonia G11 (D) and G7 (E) were determined. Of each treatment (with or without phage), the mean values of two parallel sludge systems were plotted, with the error bars indicating the standard deviations.
Variations in bacterial cell growth in two parallel sludge systems were very high, resulting in broad standard deviations at each time point (Fig. 5). This is not unexpected given the heterogeneous nature of the complex microbial populations in sludge matrix. An independent duplication of the experiment was conducted using a different batch of activated sludge as the matrix. Despite the inherent variability that arises when utilizing such a mixed test matrix, phage treatment suppressed Gordonia host levels compared to non-treated samples (Supplementary Fig. S2).
The level of recoverable phage in the sludge systems was also assayed at each time point by pfu-based titration of both phage-treated and untreated samples, on lawns of host G11 and G7 (Fig. 6). Phage was undetected (<10 pfu/ml, 10 pfu/ml is the detection limit) in sludge samples inoculated with Gordonia hosts G11 and G7 without phage treatment. While the phage-treated sludge samples initially had high levels of phage, phage titers showed consistent declines over the course of the experiments. Though a decrease in recovered phage titers was observed over time, phages against G11 and G7 could still be recovered (≥10 pfu/ml) up to six days after initial inoculation (Fig. 6A,B, respectively).

Recoverability of phage against Gordonia in the laboratory-scale activated sludge systems.
Gordonia strains G11 and G7 were inoculated to activated sludge with (empty mark) or without (filled mark) phage cocktail treatment. Mean levels of phage against host G11 (A) and host G7 (B), in two parallel sludge systems were plotted with error bars indicating standard deviations.
Finally, the morphology and settling property of sludge floc were compared between the freshly collected sludge at the start of the experiment (day zero), as well as phage-treated and untreated control sludge at the end of the experiment (day nine) (Table 4). At the end of phage treatment, both control and phage-treated sludge had significantly reduced amount of filaments around flocs compared to the freshly collected sludge. The freshly collected sludge at the start of the experiment and the phage-treated sludge at the end of the experiment had SVI values of 75.3 and 97.3 ml/g, respectively. These SVI values were higher than the SVI value of the untreated control sludge at the end of the experiment (54.9 ml/g). After sludge settling, the supernatants above the solids layer were collected and tested for foaming potential using the Alka-Seltzer® foaming test. The supernatant of the control sludge without phage treatment exhibited a higher foaming potential, evident by the greater maximum foaming volume and longer foam collapse time, compared to the phage-treated sludge and fresh sludge (Table 4). This is possibly caused by the high turbidity of the supernatant due to poor sludge settling of untreated sludge.
Discussion
Foaming is a common and ubiquitous problem in sludge management resulting in elevated costs and reduced operation efficiencies. Foaming is frequently the result of overgrowth of nocardioform filamentous bacteria, including Nocardia, Gordonia, Rhodococcus and Corynebacterium spp. Though currently unidentified factors could determine the foaming process, there is evidence that Gordonia is commonly the predominant genus within the foaming-associated mycolata15,18,19. Current control methods include mechanical removal of foam, the use of chemical defoamers such as silicone sprays and polypropylene glycols and implementation of biological control through supporting the growth of non-foaming bacteria. An improved foaming control methodology must be easily incorporated into current waste treatment protocols, be non-toxic and affect only the target microorganisms while leaving non-target species intact. These control criteria might be met by the use of phage. Phages are already a normal constituent of the wastewater microcosm and are generally host–specific. Phage application does not usually completely eliminate the target host, which is advantageous as the target filamentous bacteria at a lower level are a necessary component in promoting floc-forming and proper sludge settling during activated sludge treatment process2. There are, however, challenges in the development of phage-based foaming control agents. Propagation of the necessary phage isolates may be problematic because some of the target filamentous bacteria associated with sludge foaming have been reported to be unculturable or extremely difficult to grow in laboratory settings5,6,7. Additionally, the full extent of diversity of bacteria responsible for the foaming remain to be discovered15. Finally, phage preparations have to be shown to be effective in the mixed chemistries and microflora of activated sludge, rather than just in defined culture media.
The approach used here for isolating foaming-associated filamentous bacteria was to culture activated wastewater sludge on minimal media with defined carbon sources, in order to reduce the growth of rapidly dividing bacteria and allow microcolonies of filamentous bacteria to form. Sodium acetate and glucose generally were better for growth of filamentous bacteria, compared to other carbon sources tested. Isolating and culturing different morphotypes of foaming-associated filamentous bacteria from wastewater foams is challenging and the currently acceptable identification of filamentous bacteria was thus mainly conducted in situ, using methods based on their in situ morphological features and/or molecular FISH method using 16s rRNA-based DNA probes16. These results suggest a feasible approach for isolating Gordonia and/or other filamentous bacteria from wastewater sludge samples.
Using the isolated Gordonia strains as hosts, phages isolated from wastewater were able to control Gordonia levels in defined media (data not shown). Moreover, application of these phages resulted in repeatable, significant suppression of Gordonia levels in activated sludge conditions. This is surprising, given the considerable diversity and species richness observed in the micro- and macro-organism community of the activated sludge, with different organismal groups responsible for complex functions including floc-forming, phosphorus removal, nitrite oxidation and denitrification3,7. In such a heterogeneous and complex background consisting of viruses, bacteria, protozoa and metazoans, experiment-to-experiment variations are to be expected. Following immediate inoculation into the mixed sludge bioreactors, the host Gordonia values fluctuated at the beginning of the experiment (day zero to day one) (Fig. 5), possibly reflecting the stage required for the exogenously added Gordonia to be equilibrated into the sludge microbial system. Upon Gordonia and phage inoculation (day zero), a high level of Gordonia G7 was observed in phage-treated system compared to the control. This possibly reflects the rapid initial phage lysis and release of G7 genomic DNA, which resulted in efficient Gordonia DNA extraction and thus increased detectable quantities of Gordonia 16S rRNA genes. Despite the fluctuations at the start of the experiment, the effect of phage-treatment on Gordonia populations in the activated sludge was reproducibly detectable between day five and day nine. In these experiments, the level of indigenous background Gordonia sp. G11-like organisms was below the detection limit. In addition, there was very low level of indigenous Gordonia sp. G7-like organisms in the fresh sludge. The phage applied to the sludge system may have had a killing effect on both the indigenous and the exogenously inoculated Gordonia hosts. This study provides information on phage-Gordonia population dynamics in real activated sludge on a laboratory scale, conditions that more closely mimic the real world treatment systems compared to efficacy studies carried out in defined cultures33,36.
In addition to controlling Gordonia levels in activated sludge, phage-treated sludge at the end of the experiment showed better settling properties compared to the control. Compared to the continuous-flow aeration reactors, which resemble continuous biological enrichment cultures, the activated sludge aeration systems in this study were aerated static cultures. Compared to the freshly collected sludge, both the control and phage-treated sludge systems at the end of the experiment showed only small amount of filaments around less compact flocs. This suggested the poor health of flocs after prolonged aeration (nine days) at static culturing mode. Nevertheless, phage treated sludge had a sludge volume index (SVI) of 97.3 ml/g, which falls in the range of what is typically expected in a healthy sludge aeration basin (70–150 ml/g). SVI is an indicator best characterizing sludge settling properties. A SVI of greater than approximately 150 ml/g is often classified as bulking, while a SVI of less than approximately 70 ml/g can leave behind a turbid supernatant, the condition known as “pin-point floc”40. Compared to the control sludge with a low SVI (54.9 ml/g), the phage treated sludge at the end of the experiment had lower foaming potential in its supernatant after settlement, indicating more complete sludge settlement.
In conclusion, this study has demonstrated that phages applied for bio-control can survive during sludge aeration. Moreover, phages exhibited the capability for suppressing Gordonia hosts in real sludge and improving sludge settling property. Taken together, these results indicate significant potential for phage application in controlling Gordonia-associated foaming and bulking during wastewater treatment.
Materials and Methods
Isolation and identification of filamentous bacteria from wastewater foams
Activated sludge foam samples were collected from a local municipal wastewater treatment plant. After dispersing the flocs using low energy sonication in an ultrasonic water bath, samples were diluted and plated on Brain Heart Infusion (BHI, Difco) agar and minimal salt medium base (0.5 g/l (NH4)2SO4, 0.01 g/l Ca(NO3)2, 0.05 g/l K2HPO4, 0.05 g/l MgSO4·7H2O, 0.05 g/l KCl, 0.1 g/l CaCO3) supplemented with different carbon sources (0.15 g/l sodium acetate, sucrose, glucose, or sodium succinate). The plates were incubated at 30 °C and were examined daily under a phase contrast microscope at 100–400× magnification over one month for micro-colony development. Bacteria exhibiting filamentous growth were picked using a fine needle. These strains were purified and routinely maintained on BHI agar at 30 °C.
The identity of each isolate was determined by amplification of the 16S rDNA genes using the primer set 16S.F (5′-GGTGAGTAACACGTGGGTGA-3] and 16S.R (5′-GGGGTCGAGTTGCAGACC-3′) and sequencing the resulting ~1200 bp amplicon. The 16S rDNA sequences were analyzed via the Ribosomal Database Project (RDP) 10 suite (rdp.cme.msu.edu). The closest three matching type strain sequences for each isolate were determined by the RDP SeqMatch tool41. Sequences were aligned with ClustalX v2.1 and a neighbor joining tree was generated with gap positions excluded42. Strains were assigned at the species level based on >99.5% sequence identity to a known bacterial strain as determined from the sequence alignment. Strains with <99.5% identity or >99.5% identity to more than one species were assigned at the genus level only. The tree was visualized and rendered with Archaeopteryx v0.95743.
Phage isolation and host range determination
Foam samples collected from a local wastewater plant were centrifuged (10,000 × g, 10 min) and the filtered supernatants (through 0.22 μm filters) were mixed 1:1 with 2× BHI for phage enrichment. Phage detection and preparation were performed using BHI as top agar following standard methods44. BHI (0.5×, ~pH 6.5) was routinely used as buffer to suspend phage. The host range of phages was assessed by spotting 10 μl of lysates at routine test dilutions (RTD) onto lawns of individual hosts. The RTD of phage was defined as the last 10-fold dilution that developed confluent clearing when spotted onto a lawn of the original enrichment host.
TEM
Transmission electron microscopy (TEM) was performed by diluting lysates 1:1 to 4:1 with TEM buffer (20 mM NaCl, 10 mM Tris-HCl pH 7.5, 2 mM MgSO4) and applying to 10–15 nm carbon films by the Valentine method45. Images were generated as previous described31.
Phage genome sequencing and annotation
Phages were sequenced to ~23-fold coverage by pyrosequencing (Roche/454 Life Sciences, Branford, CT) following the protocols described previously46. Gap closure was completed by sequencing PCR products generated by the amplification of gap regions. The genomic terminal structure and end sequences were determined based on terminase homology and genome circular assembly, or based on direct sequencing of the termini using phage genomic DNA as template and self-ligating of phage genomic DNA followed by PCR and sequencing of the PCR product46. Protein-coding genes were initially predicted using Genemark.hmm47 and manually edited in Artemis48. Predicted proteins were searched against the GenBank nr database using BLASTp49. Conserved domains were detected with InterProScan version 4.7 run locally50. Protein localization was determined by analysis with TMHMM 2.0 (http://www.cbs.dtu.dk/services/TMHMM). Phage genome maps were rendered with DNA Master (http://cobamide2.bio.pitt.edu/computer.htm).
Nucleotide sequence accession number
The 16S rDNA sequences of the Gordonia isolates, were deposited in the GenBank database under the accession numbers KR067658 (G1), KR067659 (G4), KR067660 (G5), KR067661 (G7), KR067662 (G8), KR067663 (G10), KR067664 (G11). Accession numbers for phage genome sequences are: KP790008 (GordTnk2), KP790009 (Gmala1), KP790010 (GordDuk1), KP790011 (Gsput1).
Phage efficacy test in lab-scale activated sludge systems
Activated sludge freshly collected from a local wastewater plant (College Station, TX) was divided into four parallel one-liter Pyrex® ProCulture™ Spinner Flask bioreactors (4502-series, Corning) with central magnetically driven impeller assembly and two dual angled sidearm fittings. Continuous aeration in the form of filtered sterile air was introduced via longer tubes of both side arm fittings to the bottom of the bioreactors and the head space air venting was achieved via the shorter tubes of the side arm fittings. The submerged air bubbling, coupled with magnetically driven stirring of the culture, was used to supply adequate aeration to the activated sludge. Each bioreactor contained 500 ml of activated sludge. A portion (~150 ml) of the clear water phase was removed, to compensate for the volume increase due to bacteria and phage inoculation.
The bioreactors were inoculated with host bacteria to a final level of 5 × 107 cfu/ml. This was achieved by application of 25 ml of a 1 × 109 cfu/ml fresh preparation of each strain (G7 and G11). Two hours after Gordonia inoculation, 100 ml phage cocktail was applied to two of the four bioreactors. The final concentration of phage in the phage treated bioreactors was 6.5 × 108, 9.5 × 108, 3.7 × 108 and 1.0 × 109 pfu/ml for phages Gsput1, GordTnk2, Gmala1, GordDuk1, respectively. This corresponded to approximate phage treatment MOI of each phage of 5–15. Blank phage buffer (0.5× BHI, 100 ml) was inoculated into the other two bioreactors to serve as non-phage controls. Fresh activated sludge alone (without Gordonia or phage inoculation) was tested in parallel. At the start of the experiment and each day after (up to nine days), aliquots (1.5 ml each) of well-mixed sludge samples were taken aseptically using a syringe fitted through sidearm fittings and were used for phage titering and DNA isolation. The experiment was carried out independently twice, each time using freshly collected sludge. At the end of each of experiment, sludge testing was conducted as described below.
Quantification of Gordonia and total bacteria in activated sludge experiments using real time PCR
Total DNA was isolated from sludge using the Mo Bio UltraClean™ Microbial DNA Isolation kit (Mo Bio Laboratories, CA) utilizing bead-beating step. For Gordonia strains G7 and G11 quantification, the following primers and TaqMan probes targeting specific 16sRNA regions were designed in this study: Gordonia sp. strain G7, G7.F (5′- CTGGGAAACTGGGTCTAATAC); G7.R (5′- CATCCCAAACCGCAAAAG); G7.P (5′-TTCCACCACAAGACATGCATCCTGA); Gordonia sp. strain G11, G11.F (5′-CTGGGAAACTGGGTCTAATAC); G11.R (5′- CATCCCTAACCGCAAAAG); G11.P (5′- TCCACAAATCCCCATGCGAGGAA). For quantification of total bacteria, 16sRNA primers/probes reported previously were used: BacT.F (5′-TCCTACGGGAGGCAGCAGT); BacT.R (5′-GGACTACCAGGGTATCTAATCCTGTT), BacT.P (5′- CGTATTACCGCGGCTGCTGGCAC)51. PCR were performed in 384-well plates using a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) and the results were analyzed using SDS v2.3 (Applied Biosystems). For validating and calibrating the G7 and G11 primer/probe sets, standard curves were generated in the range of 0.025−50,000 pg total DNA per reaction, using G7 and G11 genomic DNA, respectively. For calibrating total bacteria primer/probe set, G11 genomic DNA was used to generate standard curve. Quantification efficiencies, detection range and Ct were calculated from the standard curves. The optimal sludge DNA input level was determined to be 10 ng based on the Ct values for G7, G11 and total bacteria quantification in the DNA input range of 1–100 ng. To test real samples, Each 20-μl reaction mixture contained 10 μl of 2× TaqMan Universal Master mix II with UNG (Applied Biosystems), 10 ng total DNA, 625 nM each of forward and reverse primers (Integrated DNA Technologies, Coralville, IA) and 250 nM of probe (5′-/56-FAM/-/ZEN/-/3IABkFQ/-3′ probe, Integrated DNA Technologies). The thermal profile consisted of 50 °C for 2 min, 95 °C for 10 min and 40 cycles of 95 °C for 15 sec, 60 °C for 60 sec. All samples were measured in duplicate in each assay. Standard curves (generated using G11 or G7 genomic DNA) and negative controls were included in each PCR run. The average size of the Gordonia genome was estimated to be 5.2 Mbp, based on the sequenced genome of G. bronchialis DSM43247, NC_013441). By comparison of the fluorescence generated by each activated sludge sample with standard curves, the quantities of bacterial and Gordonia 16s rRNA genes of activated sludge samples were interpreted as Gordonia genome equivalents per 10 ng of extracted DNA used in PCR reaction. This value was then adjusted for the total DNA extracted from 1.5 ml sludge and the final result was expressed as Gordonia genome equivalents per ml of sludge.
Activated sludge testing
Floc morphology was examined using a phase-contrast microscope (Fisher Scientific MicromasterTM, Pittsburgh, PA) under 1000× magnification. The total suspended solids concentration of sludge was determined by filtering and drying sludge aliquots following the standard method52. The sludge volume index (SVI) was determined by transferring the sludge to a 1 liter graduated cylinder and allowing the sludge to settle undisturbed for 30 min. The volume occupied by the settled sludge was recorded and used to calculate the SVI (ml/g) = settled sludge volume (ml/l)/total suspended solids (g/l). After sludge settlement, the supernatant above the settled layer was subjected to the Alka-Seltzer® foaming potential test as described previously1.
Additional Information
How to cite this article: Liu, M. et al. Bacteriophages of wastewater foaming-associated filamentous Gordonia reduce host levels in raw activated sludge. Sci. Rep. 5, 13754; doi: 10.1038/srep13754 (2015).
References
Jenkins, D., Richard, M. G. & Daigger, G. T. Manual on the causes and control of activated sludge bulking, foaming and other solids separation problems, Edn. 3. (CRC Press, Boca Raton; 2004).
Martins, A. M., Pagilla, K., Heijnen, J. J. & van Loosdrecht, M. C. Filamentous bulking sludge–a critical review. Water Res 38, 793–817 (2004).
Wagner, M. et al. Microbial community composition and function in wastewater treatment plants. Antonie Van Leeuwenhoek 81, 665–680 (2002).
Petrovski, S. et al. An examination of the mechanisms for stable foam formation in activated sludge systems. Water Res 45, 2146–2154 (2011).
Eikelboom, D. H. Filamentous organisms observed in activated sludge. Water Res 9, 365–388 (1975).
Eikelboom, D. H. Identification of filamentous organisms in bulking activated sludge. Prog. Water Tech. 8, 152–161 (1977).
Elkelboom, D. H. & Geurkink, B. Filamentous micro-organisms observed in industrial activated sludge plants. Water Sci Technol 46, 535–542 (2002).
van der Waarde, J. et al. Molecular monitoring of bulking sludge in industrial wastewater treatment plants. Water Sci Technol 46, 551–558 (2002).
Kragelund, C. et al. Ecophysiology of different filamentous Alphaproteobacteria in industrial wastewater treatment plants. Microbiology 152, 3003–3012 (2006).
Kragelund, C. et al. Identity, abundance and ecophysiology of filamentous bacteria belonging to the Bacteroidetes present in activated sludge plants. Microbiology 154, 886–894 (2008).
Speirs, L., Nittami, T., McIlroy, S., Schroeder, S. & Seviour, R. J. Filamentous bacterium Eikelboom type 0092 in activated sludge plants in Australia is a member of the phylum Chloroflexi. Appl Environ Microbiol 75, 2446–2452 (2009).
Ju, F., Guo, F., Ye, L., Xia, Y. & Zhang, T. Metagenomic analysis on seasonal microbial variations of activated sludge from a full-scale wastewater treatment plant over 4 years. Environ Microbiol Rep 6, 80–89 (2014).
Guo, F. & Zhang, T. Profiling bulking and foaming bacteria in activated sludge by high throughput sequencing. Water Res 46, 2772–2782 (2012).
Zhang, T., Shao, M. F. & Ye, L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J 6, 1137–1147 (2012).
Guo, F., Wang, Z. P., Yu, K. & Zhang, T. Detailed investigation of the microbial community in foaming activated sludge reveals novel foam formers. Sci Rep 5, 7637 (2015).
Nielsen, P. H., Kragelund, C., Seviour, R. J. & Nielsen, J. L. Identity and ecophysiology of filamentous bacteria in activated sludge. FEMS Microbiol Rev 33, 969–998 (2009).
Goodfellow, M., Alderson, G. & Chun, J. Rhodococcal systematics: problems and developments. Antonie Van Leeuwenhoek 74, 3–20 (1998).
de los Reyes, M. F., de los Reyes, F. L., 3rd, Hernandez, M. & Raskin, L. Quantification of Gordona amarae strains in foaming activated sludge and anaerobic digester systems with oligonucleotide hybridization probes. Appl Environ Microbiol 64, 2503–2512 (1998).
Kragelund, C. et al. Ecophysiology of mycolic acid-containing Actinobacteria (Mycolata) in activated sludge foams. FEMS Microbiol Ecol 61, 174–184 (2007).
Seka, M. A., Hammes, F. & Verstraete, W. Predicting the effects of chlorine on the micro-organisms of filamentous bulking activated sludges. Appl Microbiol Biotechnol 61, 562–568 (2003).
Withey, S., Cartmell, E., Avery, L. M. & Stephenson, T. Bacteriophages–potential for application in wastewater treatment processes. Sci Total Environ 339, 1–18 (2005).
Thomas, J. A., Soddell, J. A. & Kurtboke, D. I. Fighting foam with phages? Water Sci Technol 46, 511–518 (2002).
Ewert, D. L. & Paynter, M. J. Enumeration of bacteriophages and host bacteria in sewage and the activated-sludge treatment process. Appl Environ Microbiol 39, 576–583 (1980).
Khan, M. A., Satoh, H., Katayama, H., Kurisu, F. & Mino, T. Bacteriophages isolated from activated sludge processes and their polyvalency. Water Res 36, 3364–3370 (2002).
Summers, W. C. Bacteriophage therapy. Annu Rev Microbiol 55, 437–451 (2001).
Sulakvelidze, A., Alavidze, Z. & Morris, J. G., Jr. Bacteriophage therapy. Antimicrob Agents Chemother 45, 649–659 (2001).
Matsuzaki, S. et al. Bacteriophage therapy: a revitalized therapy against bacterial infectious diseases. J Infect Chemother 11, 211–219 (2005).
Merril, C. R., Scholl, D. & Adhya, S. L. The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov 2, 489–497 (2003).
Hatfull, G. F. Mycobacteriophages: genes and genomes. Annu Rev Microbiol 64, 331–356 (2010).
Hatfull, G. F. et al. Comparative genomic analysis of 60 Mycobacteriophage genomes: genome clustering, gene acquisition and gene size. J Mol Biol 397, 119–143 (2010).
Summer, E. J. et al. Genomic and functional analysis of Rhodococcus equi phages ReqiPepy6, ReqiPoco6, ReqiPine5 and ReqiDocB7. Appl Environ Microbiol 77, 669–83 (2011).
Kotay, S. M., Datta, T., Choi, J. & Goel, R. Biocontrol of biomass bulking caused by Haliscomenobacter hydrossis using a newly isolated lytic bacteriophage. Water Res 45, 694–704 (2011).
Petrovski, S., Seviour, R. J. & Tillett, D. Prevention of Gordonia and Nocardia stabilized foam formation using bacteriophage GTE7. Appl Environ Microbiol 77, 7864–7 (2011).
Petrovski, S., Seviour, R. J. & Tillett, D. Genome sequence and characterization of the Tsukamurella bacteriophage TPA2. Appl Environ Microbiol 77, 1389–1398 (2011).
Khairnar, K., Pal, P., Chandekar, R. H. & Paunikar, W. N. Isolation and characterization of bacteriophages infecting nocardioforms in wastewater treatment plant. Biotechnol Res Int 2014, 151952 (2014).
Petrovski, S., Seviour, R. J. & Tillett, D. Characterization of the genome of the polyvalent lytic bacteriophage GTE2, which has potential for biocontrol of Gordonia-, Rhodococcus- and Nocardia-stabilized foams in activated sludge plants. Appl Environ Microbiol 77, 3923–3929 (2011).
Gill, J. J. & Young, R. Therapeutic Applications of Phage Biology: History, Practice and Recommendations. (Caister Academic Press, Norfolk, UK; 2011).
Pope, W. H. et al. Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution. PLoS One 6, e16329 (2011).
Hatfull, G. F. Complete genome sequences of 138 mycobacteriophages. J Virol 86, 2382–2384 (2012).
Palm, J. C., Jenkins, D. & Parker, D. S. Relationship between organic loading, dissolved oxygen concentration and sludge settleability in the completely-mixed activated sludge process. Journal of Water Pollution Control Federation 52, 2484 (1980).
Cole, J. R. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37, D141–145 (2009).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Han, M. V. & Zmasek, C. M. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics 10, 356 (2009).
Adams, M. H. Bacteriophages. (Interscience Publishers, New York; 1959).
Valentine, R. C., Shapiro, B. M. & Stadtman, E. R. Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 7, 2143–2152 (1968).
Summer, E. J. Preparation of a phage DNA fragment library for whole genome shotgun sequencing. Methods Mol Biol 502, 27–46 (2009).
Lukashin, A. V. & Borodovsky, M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res 26, 1107–1115 (1998).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res 37, D211–215 (2009).
Nadkarni, M. A., Martin, F. E., Jacques, N. A. & Hunter, N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266 (2002).
Eaton, A. D., Clesceri, L. S., Rice, E. W. & Greenberg, A. E. Standard methods for the examination of water and wastewater, Edn. 21. (American Public Health Association, Washington DC; 2005).
Acknowledgements
This work is supported by NSF SBIR Award No. IIP-0945913, AgriLife Research, Center for Phage Technology and Texas A&M University. We thank Greg Wall and other staff at College Station wastewater treatment plant for collecting samples. We thank staff at wastewater treatment plants in Bryan, Navasota and Willis, TX.
Author information
Authors and Affiliations
Contributions
M.L. and E.J.S. contributed to sample collection, experimental design and conduct, data analysis and manuscript preparation. J.J.G. and R.Y. contributed to the data analysis and manuscript revision.
Ethics declarations
Competing interests
M.L. and E.J.S. work for Ecolyse Inc. J.J.G. and R.Y. declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, M., Gill, J., Young, R. et al. Bacteriophages of wastewater foaming-associated filamentous Gordonia reduce host levels in raw activated sludge. Sci Rep 5, 13754 (2015). https://doi.org/10.1038/srep13754
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13754
This article is cited by
-
Two novel bacteriophages isolated from the environment that can help control activated sludge foaming
Folia Microbiologica (2024)
-
Bacteriophages in wastewater treatment: can they be an approach to optimize biological treatment processes?
Environmental Science and Pollution Research (2022)
-
Microbial predation accelerates granulation and modulates microbial community composition
BMC Microbiology (2021)
-
Long-run bacteria-phage coexistence dynamics under natural habitat conditions in an environmental biotechnology system
The ISME Journal (2021)
-
Prokaryotic viruses impact functional microorganisms in nutrient removal and carbon cycle in wastewater treatment plants
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
