
Abstract
Although technology has triumphed in facilitating routine genome sequencing, new challenges have been created for the data-analyst. Genome-scale surveys of human variation generate volumes of data that far exceed capabilities for laboratory characterization. By incorporating functional annotations as predictors, statistical learning has been widely investigated for prioritizing genetic variants likely to be associated with complex disease. We compared three published prioritization procedures, which use different statistical learning algorithms and different predictors with regard to the quantity, type and coding. We also explored different combinations of algorithm and annotation set. As an application, we tested which methodology performed best for prioritizing variants using data from a large schizophrenia meta-analysis by the Psychiatric Genomics Consortium. Results suggest that all methods have considerable (and similar) predictive accuracies (AUCs 0.64–0.71) in test set data, but there is more variability in the application to the schizophrenia GWAS. In conclusion, a variety of algorithms and annotations seem to have a similar potential to effectively enrich true risk variants in genome-scale datasets, however none offer more than incremental improvement in prediction. We discuss how methods might be evolved for risk variant prediction to address the impending bottleneck of the new generation of genome re-sequencing studies.
Similar content being viewed by others

Introduction
Complex diseases are caused by the interplay of many genetic variants and the environment and represent a considerable health burden. Genome-wide association studies (GWAS) have had success in identifying some genetic risk factors involved in complex diseases such as inflammatory bowel disease1 and schizophrenia2. Interrogating the entire genome, exome or even selected genes through next generation sequencing technologies have also identified further risk variants3,4,5,6. However, more disease-associated variants, hereafter called risk variants or hits, remain to be discovered. Some risk variants are difficult to detect by current techniques due to limited sample sizes and low effect size of the variants. In silico methodologies that integrate evidence over multiple data sources have the potential to unearth some of these risk variants in a cost-effective manner. The novel risk variants that are identified will help illuminate the genetic risk factors involved in complex diseases, which in turn could lead to earlier or more accurate diagnoses and the development of personalized treatment options.
Risk variants show enrichment in functional annotations, such as DNase I hypersensitive sites, transcription factor binding sites and histone modifications (for example7,8,9). Several groups have gone further with the results of enrichment by incorporating functional annotations as predictor variables in statistical learning frameworks to prioritize genetic variants for further study10,11,12. These statistical learning algorithms use the functional annotations to define a model that provides some measure of whether a variant is likely to increase the risk of manifesting a complex trait. However, understanding the relative merits of these approaches requires a thorough investigation into which statistical learning algorithm and/or which combination of functional annotations most effectively identifies novel risk variants.
There are many aspects to consider in the statistical learning framework (Supplementary Fig. 1). The genetic data input consists of both known risk variants and corresponding control variants (those with no evidence for risk effect); the classifier is used to discriminate between the two. Known risk variants may be identified from sources, such as the National Health Genome Research Institute (NHGRI) GWAS Catalogue13, the ClinVar database14 and the Human Gene Mutation Database (HGMD)15. In addition, the variants can be simulated; for example, Kircher et al. used an empirical model of sequence evolution with local adjustment of mutation rates11. In this way, the simulated variants would contain de novo pathogenic mutations. The goal of these methods is to identify disease-causing variants, but their application can differ depending on whether the data under consideration consist of densely mapped variants, as in sequence data, or coarsely mapped variants, as in GWAS data. The use of different classifiers has the effect of refining the goal, in that coarsely mapped variants may tag other variants in high linkage disequilibrium and so the functional characteristics of these other variants should be taken into account. The methods we investigate have been applied to both types of data16,17.
With regard to the functional annotations, some come from experimental procedures while others are predicted computationally. Examples include genomic and epigenomic annotations that can be incorporated from various online browsers and collections such as the Ensembl Variant Effect Predictor (VEP)18 and the Encyclopedia of DNA Elements (ENCODE) Project19. Whether a variant is assigned the annotations that can be attributed to itself only or to other variants with which it is in linkage disequilibrium can also refine the goal of the method.
Finally, there are numerous statistical learning algorithms from which to choose. These algorithms must be able to handle the features of the functional data: correlations among predictor variables and a large quantity of both samples and predictor variables.
In this paper, we compared the performance of three published methods that differ in annotation set, algorithm and genetic variants, including the classifier: a regularized regression called elastic net from Gagliano et al. (14 annotations)10, a modified random forest from Ritchie et al. (174 annotations)12 called GWAVA and a support vector machine from Kircher et al. (949 annotations, expanded from 63 unique annotations) called CADD11. These three papers describe algorithms capable of incorporating a large number of genetic variants labeled with multiple functional annotations and can output a prediction score for each variant; hence, they are highly comparable. Although other methods exist to prioritize genetic risk variants, such as through the use hierarchical Bayesian analysis20,21, these require genetic association statistics for each variant for prioritization and thus were beyond the scope of the comparisons in this paper. We investigate nine model types: combinations of the three different statistical learning algorithms and the three different functional annotation sets (summarized in Table 1). All model types were created for different classifications of hits: the NHGRI GWAS Catalogue13 and the Human Gene Mutation Database (HGMD)15.
Models based on GWAS data can be tested effectively in current data (we apply those models to the schizophrenia GWAS from the Psychiatric Genomics Consortium).
Results
Our primary analysis used the NHGRI GWAS Catalogue as the classifier. Risk variants/hits were defined as those variants present in the NHGRI GWAS Catalogue (www.genome.gov/gwastudies, downloaded on August 7, 2014)13 with a p-value of equal to or less than the accepted threshold for genome-wide significance, 5 × 10−8. A subset of non-hits (that are not in high linkage disequilibrium with the hits) was selected from common GWAS arrays for comparability. For the three annotation sets described above, when working with different classifiers some rare annotations have no variability and hence were not used to build the model. In this analysis none of the 14 annotations from Gagliano et al. were invariable, three of the 174 annotations from Ritchie et al. were invariable and 509 of the 949 annotations from Kircher et al. were invariable. An independent test set was used to determine accuracy of the models for discriminating hits from non-hits based on the predictive score output from each model. These results are presented below.
Area under the ROC curve
All the models had similar accuracy as demonstrated by the area under the curve (AUC) in the test set data (Table 2). Models using Kircher et al.’s annotations produced slightly higher AUCs compared to the other two annotation sets for the elastic net and random forest algorithms. In particular the combination of elastic net and Kircher et al.’s annotations was the only model that produced an AUC with confidence intervals that do not overlap with any of the other models.
The AUC results for the training set were also computed to investigate whether the models were over-fit; that is to say, whether the training set AUC is much higher than the test set AUC. We found that for the Ritchie et al. and Kircher et al. annotation sets, the random forest models with node size equal to one were prone to over-fitting. For instance, for the random forest model based on the Ritchie et al. annotations, the test set and training set AUCs were 0.687 and 0.998, respectively (further data available on request). The over-fitting in the random forest models was solved when the minimum node size was set to 10% of the total sample size. Therefore only the random forest models with the minimum node size equal to 10% of the data are presented in Table 2 and discussed further in the results. These results highlight the importance of ensuring that appropriate parameters are chosen for the algorithms.
Density and distribution of prediction scores
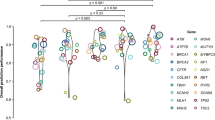
Violin plots were constructed by plotting the prediction scores for hits (risk variants) and non-hits separately in order to visualize how well the two classes separated (Fig. 1 and Supplementary Table 1a). The two models with the best AUCs (Kircher et al. annotations with elastic net (0.71) and with random forest (0.70)) have comparatively well separated means and relatively normal distributions. In one of the two models with the lowest AUC (Ritchie et al. annotations with support vector machine (0.64)), the median prediction score between hits and non-hits is most similar and the distribution is very skewed. Interestingly, one of the mid-range performance models, the Gagliano et al. annotations for the support vector machine (0.66) showed evidence of a multimodal distribution where one mode is more common for hits and another for non-hits. However, this effect may simply be due to the comparatively small number of annotations, which lead to a smaller number of possible scores. Generally, the models created using the Kircher et al. annotations showed the largest spread of prediction scores for both hits and non-hits. We have also reported the proportion of hits in the top versus the bottom quartiles of the prediction scores in the test set (Supplementary Table 2). In summary the violin plots show that the distributions for hits and non-hits overlapped for all models. However, we see from Supplementary Table 2 that of the variants in the top quartile of prediction scores, there are significantly more hits compared to the lower quartile for all models assessed (p < 2.2 × 10−16, chi-square test).

Violin plots showing class separation by prediction scores for the various comparisons using the GWAS Catalogue as the classifier.
Hits are variants in the GWAS Catalogue with a genome-wide significant p-value (p ≤ 5 × 10−8) and non-hits are those not present in the GWAS Catalogue, but are found on common GWAS arrays for comparison purposes. The non-scaled elastic net models are plotted. The adjusted minimum node size (10%) random forest models are plotted.
To investigate the consistency of the models we calculated pairwise correlations of the prediction scores in the test set for the various models either holding the algorithm or the annotation set constant. We found that the models with the most correlated scores were those using the Gagliano et al. annotation set. Furthermore, the degree of correlation when holding the algorithm constant, but varying the annotation set, was generally not as high as when holding the annotation set constant (Supplementary Table 3).
Feature selection within elastic net and random forest
More does not necessarily equal better as not all the annotations may be relevant to predicting risk variants. Generally, not all of the functional annotations in the annotation sets were used to create the various models. For instance of the variable features, elastic net assigned non-zero Beta coefficients to 9 out of 14 annotations, 12 out of 171 and 16 out of 432. Random forest assigned non-zero Gini importance values to all of the 14, 131 out of 171 and 239 out of 432. All of these models had similar performance in the test sets (AUCs ranging from 0.68 to 0.70 for the random forest models and 0.65 to 0.71 for the elastic net models). The results suggest that elastic net has a more stringent feature selection implementation than random forest. The support vector machine models always assigned non-zero feature weights, as support vector machine does not intrinsically perform feature selection, as does elastic net and random forest. Thus, we inputted only those annotations with a non-zero Beta coefficient from the elastic net models into the support vector machine models (see Methods).
Importance of the functional annotations
Different combinations of annotations can be used to obtain models with similar predictive accuracy. Furthermore, it is difficult to interpret the importance of the annotations for numerous reasons, some of which are discussed below.
All three annotation sets contained a mixture of binary variables and continuous variables. For Kircher et al.’s annotations, background selection (the annotation with the widest continuous scale that ranged from 0 to 1000) came up as most important for predicting the class label in the random forest model. This bias for random forest preferentially selecting annotations measured on a continuous scale has been previously described22. When making a decision at a node, continuous annotations can be used multiple times at varying cut-offs to split the data. In this way, functional annotations measured on a continuous scale are incorporated more often into the forest compared to non-continuous annotations and thus obtain higher variable importance measures22,23.
It is also difficult to interpret the variable importance measures derived from elastic net because this algorithm is not scale invariant. Using Gagliano et al.’s annotations with elastic net, we compared the models created with scaled (all annotations have a standard deviation of 1 and a mean of 0) versus non-scaled annotations. Although the AUCs for both models were nearly identical, the assigned Beta coefficients differed (Supplementary Fig. 2). When we do standardize the scale, we find that the order of importance of coefficients replicates that of the random forest model. However, standardizing a set of largely binary variables removes the effect linked to the frequency and thus skews the biological representation. So it is not clear that scaling is the best approach.
Although the focus is not about annotations we have provided details of the various importance measures in supplementary data: see Supplementary Tables 9–17 and Supplementary Figures 5–13 for the feature importance measures from all the models based on the GWAS Catalogue as the classifier. In the primary analysis transcription factor binding sites were consistently in the top three annotations for the Gagliano et al. annotations for all three algorithms, but there were no other clear patterns with regard to important annotations for the Ritchie et al. or Kircher et al. annotation sets. In summary, different annotations came up as most important for the various models regardless of predictive accuracy.
Performance for complex disease variants: Application to Schizophrenia GWAS
Various quantile-quantile plots were constructed in order to compare which models showed greater separation of the schizophrenia GWAS p-values for high scoring and low scoring functional variants. For all of the models, scores were obtained for the sub-genome-wide-significant variants (5 × 10−8 < p < 1 × 10−6) from the first round of the GWAS by the Psychiatric Genomics Consortium (PGC1)24. The PGC1 p-values were plotted on the x-axis and the p-values from the second larger round of the schizophrenia GWAS (PGC2)2 were plotted on the y-axis (Fig. 2). (The results from PGC2 were not used to train the model.) Plots were constructed where annotations were held constant but the algorithm differed. For instance, for the 14 annotations from Gagliano et al. we plotted the models from the three algorithms in one plot. Furthermore, models from the same algorithm but varying by annotation set were compared (Supplementary Fig. 3). We have also reported the proportion of hits in the top versus the bottom quartiles of the prediction scores in the test set (Supplementary Table 4). With regard to the functional annotation set, the separation of the novel associated variants from the non-associated in the sub-genome-wide-significant variants was best exhibited in the quantile-quantile plots when using either the Kircher et al. or Ritchie et al. annotation sets. Regardless of annotation set, the elastic net models consistently showed good separation. For all algorithms using either the Ritchie et al. or Kircher et al. annotations, the PGC1 sub-genome-wide-significant variants that have the highest prediction scores (within the top quartile) consistently contain a higher proportion of GWAS significant variants from the second round of the schizophrenia GWAS (p ≤ 5 × 10−8) compared to the variants that have scores in the lower quartile. The elastic net models too, regardless of annotation set, showed this pattern. Although these patterns are not all statistically significant, it is notable that the biggest positive difference comes from using the Ritchie et al. annotations with the elastic net algorithm and the most significant difference between the proportion of GWAS significant variants in the top quartile compared to the proportion in the lower quartile comes from the Kircher et al. annotations using the elastic net algorithm; (there are more variants available in the Kircher et al. model than the Ritchie et al. model). The Gagliano et al. annotations performed very poorly with both the random forest and support vector machine algorithms since the variants with low prediction scores were more likely to be hits than those with high scores. This is a result of the PGC2 hits not being enriched in two of the top annotations for the Gagliano et al. models using either the random forest or support vector machine algorithms, H3K4Me3 and H3K27Ac. In the GWAS Catalogue analysis of the variants that possess the H3K4Me3 and H3K27Ac marks, nearly 70% are hits and the remainder are non-hits. In comparison, of the PGC1 sub-genome-wide-threshold variants that possess those two annotations, only 21% are PGC2 hits and the remaining variants are non-hits.

Quantile-quantile plots of PGC1 sub-genome-wide-significant variants (5 × 10−8 < p < 1 × 10−6) stratified by prediction score for the various models based on the GWAS Catalogue classifier and plotted by PGC2 p-values.
PGC1 p-values are plotted on the x-axis and PGC2 p-values are plotted on the y-axis. Models grouped by annotation set: Gagliano et al. (a) Ritchie et al. (b) and Kircher et al. annotations (c). The lower quartile genetic variants are those PGC1 sub-genome-wide-significant variants that were assigned the lowest prediction scores (in the first quartile) and the top quartile variants are those with the highest prediction scores (in the fourth quartile).
The results for the application to the schizophrenia GWAS did not always reflect the AUCs from the training data. For instance, a poor performing model in terms of AUC based on the test set, elastic net with the Ritchie et al. annotations, performed well in the GWAS application. All in all, the accuracy of the resulting models should be assessed by various means, including (but not limited to) theoretical models such as the ROC curve, as well as empirical approaches such as applying the model using data from one study and evaluating its performance on independent data with gold standard answers.
HGMD Analysis
In an attempt to apply the algorithms and annotation set combinations to whole genome sequencing data and indeed fine-mapping studies, rather than just GWAS, a different classifier was used to identify hits and non-hits, the Human Gene Mutation Database (HGMD). We conducted two analyses with subsets of the public release of HGMD. In the first, we took all the variants (single nucleotide polymorphisms) in HGMD and chose controls that fell within a kilobase of either side from the HGMD variant. In this analysis one of the 14 annotations from Gagliano et al. was invariable, eight of the 174 annotations from Ritchie et al. were invariable and 396 of the 949 annotations from Kircher et al. were invariable. Secondly, models based on the subset of non-exonic HGMD variants and non-exonic control variants were assessed. This second set of models was created in an effort to overcome the ascertainment bias inherent in HGMD related to genes. In this analysis two of the 14 annotations from Gagliano et al. were invariable, 16 of the 174 annotations from Ritchie et al. were invariable and 756 of the 949 annotations from Kircher et al. were invariable.
The models for the analysis using all of the HGMD variants using either the Ritchie et al. or Kircher et al. annotations had high predictive accuracy (Table 3). The AUCs for the non-exonic HGMD analysis were more comparable to the ones obtained for the primary analysis using the GWAS Catalogue as the classifier (Table 4), but again the annotations from Ritchie et al. and Kircher et al. performed better.
Similar to the analysis using the GWAS Catalogue as the classifier, for the HGMD analysis models the features that came up as most important tended to vary depending on the algorithm and are difficult to interpret. It is however notable that genic annotations featured highly (see Supplementary Tables 18–26 and Supplementary Figures 14–22). For the Gagliano et al. annotations, the top annotation (or the second most important in the case of support vector machine) was nonsynonymous SNPs. For the Kircher et al. annotations, the top annotations for the random forest and support vector machine models were related to the coding sequence or nonsynonymous SNPs. The top annotation for elastic net was CpG. For the Ritchie et al. annotations, the top two annotations were coding sequence and exon for both the random forest and support vector machine models. For elastic net, the top two annotations were donor and coding sequence. The importance of genic features is likely linked to bias in the data, which will be examined further in the Discussion.
The HGMD analysis in which only non-exonic HGMD and control variants were considered seemed to overcome this bias towards genes or positions relative to genes. Interestingly, for all algorithms, the top annotation for the Gagliano et al. annotation set was DNase I hypersensitive sites, but we caution against making biological inferences on the top annotations for the reasons outlined above (see Supplementary Tables 27–35 and Supplementary Figures 23–31).
Discussion
We found that the three algorithms assessed here, elastic net, random forest and the linear support vector machine show comparable accuracy in the GWAS test data. The Kircher et al. annotations trained using the elastic net algorithm have the highest AUC. When applied to real data, several models show the potential to prioritize novel hits, with the exception of the random forest and support vector machine models using the Gagliano et al. annotations. However, this was just one real dataset and further studies would need to be assessed to validate this conclusion. Under the conditions employed in our analysis, none of the models were over-fitted, as demonstrated by verifying that the training set AUC is similar in magnitude to that of the test set.
Furthermore, our results show that various combinations of annotations can create models with similar predictive ability when it comes to identifying risk variants from non-risk variants. One must be wary of making strong conclusions about the relevance of the annotations because of the difficulty in interpretation. The coefficients or variable importance measures are differentially affected by issues such as correlation between the attributes and whether variables are normalized (for elastic net and support vector machine). This observation makes it difficult to differentiate the predictive power of the functional annotation sets used by each study, at least in the case of GWAS risk variants.
As mentioned in the Introduction, the main goals of these methods are to identify those variants that are important for disease risk, which can be applied to identifying novel loci or for fine-mapping at previously implicated loci. The HGMD is designed to contain disease variants, whereas the GWAS Catalogue contains variants associated with disease, but those variants may only be tagging the “causal” variant. GWAS are undertaken to identify the loci containing the variant and may identify the actual causal variant but will more often identify variant in high linkage disequilibrium with the causal variant. Thus, the primary analyses in this paper (using the GWAS Catalogue) may be considered to be about identifying novel loci rather than fine-mapping and the HGMD analyses may be considered to be more about fine-mapping a specific locus. Furthermore, the Gagliano et al. method may be considered to be better suited to identifying novel loci (rather than fine-mapping) because it annotates variants on whether or not the variant itself falls into the base pair range for the functional annotation, but also if that variant has is in linkage disequilibrium (r2 > 0.8) with a variant that falls into the range. The Ritchie et al. and Kircher et al. methods annotate the variants just based on whether the variant itself falls into the base sequence for the functional annotation and do not look at their linkage disequilibrium proxies. That being said, we also performed the analyses for the Gagliano et al. annotations only considering whether the variant itself falls into the sequence for the functional annotation as an additional analysis. The resulting models had very similar accuracy to those models created when the linkage disequilibrium proxies were taken into account (data available on request).
To apply the methods in next generation sequencing data and fine-mapping studies we would ideally use risk variants identified from such studies. Unfortunately, there are not a sufficient number available. We used the HGMD to attempt to extrapolate our findings. However, we believe the high accuracies achieved for the all HGMD models (ie. not the models looking just at non-exonic variants) are driven by the inherent bias of the HGMD data, in that it is largely focused on genes. For the models using only non-exonic HGMD and control variants, the AUCs were considerably lower, with the Kircher et al. and Ritchie et al. annotation sets clearly out-performing the annotations used by Gagliano et al. Yet, this subset of HGMD is a highly derived and filtered set of variants, emphasizing the need for empirical data. The simulation employed by Kircher et al. to consider all variants, in which the functional annotations were used to differentiate between millions of high frequency human-derived alleles from the same number of simulated alleles11, showed considerable accuracy; further adaptions to this strategy may prove useful.
Compared to the corresponding elastic net or random forest models, the support vector machine models consistently produced slightly lower AUCs for the GWAS Catalogue and all HGMD analyses. This poorer performance may be attributed to the fact that we implemented the most basic kernel type for the support vector machine, a linear kernel. This kernel was chosen in an effort to be consistent with the type of kernel that was utilized by Kircher et al. and with the advantage that computational time remains comparable with the other algorithms (Supplementary Text and Supplementary Tables 4–7). However, a linear kernel may not be best to separate the data. Furthermore, as support vector machine does not intrinsically perform feature selection, we selected a subset of features with a non-zero Beta coefficient from the corresponding analysis using the elastic net algorithm. Use of another method of feature selection may have yielded different results. Our results do not necessarily suggest that the elastic net and random forest algorithms out-perform the support vector machine algorithm, since altering either the kernel type or the functional annotations in the support vector machine models may produce results comparable to the other two algorithms.
There are limitations to this comparison. For example, other statistical learning algorithms, such as a deep neural network25 and other annotation sets could be explored. Annotation sets could be phenotype specific, as there is evidence that the level of enrichment of functional information can differ depending on the subset of risk variants selected26. For instance, enrichment of disease-specific variants in the GWAS Catalogue can differ in certain cell types, for example for DNase I hypersensitive sites8.
Identifying which algorithm and/or annotations identify risk variants with the highest accuracy will help researchers develop a better understanding of the genetic factors involved in complex disease in a cost-effective manner making use of a rich set of publically available functional data. This work helps illuminate the genetic factors involved in disease by making use of existing functional data in silico. Increasing knowledge on the etiology of complex disease will allow for earlier or better diagnoses and the development of personalized treatment and novel therapies.
Methods
We explored the utility of each of the three algorithms with each of the three functional annotation sets in order to attribute performance differences to the algorithm and/or annotations. A total of nine model types were created.
In the primary analysis, the set of risk variants used for training all the models were based on whether or not a genetic variant is a hit or a non-hit from a genome-wide association study (GWAS). Hits were defined as those variants present in the NHGRI GWAS Catalogue (www.genome.gov/gwastudies, downloaded on August 7, 2014)13 with a p-value of equal to or less than 5 × 10−8. There were 3,618 unique genetic variants that met these criteria. (Note that at the time of download the novel hits from the second phase of the schizophrenia GWAS from the Psychiatric Genomics Consortium (PGC2)2 had not yet been included.) A subset of non-hits was selected from common GWAS arrays (Affymetrix Genome-Wide Human SNP Array 6.0, the Illumina Human1M-Duo Genotyping BeadChip and the Illumina HumanOmni1-Quad BeadChip). Those non-hits in high linkage disequilibrium (r2 > 0.8) with hits were removed from the analyses.
Functional annotation sets
The data was then annotated using three distinct protocols outlined in each of the three respective papers. The variants were marked with the Gagliano et al. annotations available on the website (http://www.camh.ca/en/research/research_areas/genetics_and_epigenetics/Pages/Statistical-Genetics.aspx). Fourteen functional annotations were used by Gagliano et al., two of which were on a continuous scale (two conservation measures, PhyloP and PhastCons) and the remaining were binary, signifying the presence or absence. The binary annotations included those related to genomic context such as the presence in a gene, a splice site or a transcription start site, as well as those from the ENCODE Project19 such as three types of histone modifications and DNase I hypersensitivity. For the ENCODE data, functional annotations present in multiple cell lines were grouped together and genetic variants were annotated accordingly in a binary, present or absent, fashion. Variants were marked with an annotation if they or their linkage disequilibrium proxies fall into the base pair range of the annotation.
To annotate the variants using Ritchie et al.’s annotations, the data were entered into the online GWAVA webserver (https://www.sanger.ac.uk/resources/software/gwava/). Ritchie et al. investigated 174 functional annotations, some binary and others continuous. They also used ENCODE Project tracks including those investigated in Gagliano et al. but not necessarily coded as presence or absence. For instance, for transcription factor binding sites, the number of cell types in which the site was present was used as the annotation. Additionally, variation such as mean heterozygosity and genic and sequence contexts were included. Variants were marked with an annotation if they fall into the base pair range of the annotation.
To obtain Kircher et al.’s annotations, the data were entered into the online CADD webserver (http://cadd.gs.washington.edu). However, Kircher et al. also imputed missing values, expanded categorical variables, added indicator variables and included interaction terms. Martin Kircher provided scripts to run on the webserver output to prepare our dataset in accordance with the complete protocol. Kircher et al. looked at 63 unique functional annotations, which totaled to 949 once the categorical variables were expanded and the indicator variables and interaction terms were included. A mixture of continuous, categorical and binary functional annotations was included. Similar annotations to those used by Gagliano et al. and/or Ritchie et al. were included, such as ENCODE Project annotations and genic context. Additionally, data from online variant prediction programs (e.g. Sift27 and PolyPhen28) were incorporated. Variants were marked with an annotation if they fall into the base pair range of the annotation.
Statistical learning algorithms
The variants were randomly divided; 60% was used for training the models and the remaining 40% was reserved for testing. Elastic net is a regularized logistic regression and those models were constructed using the glmnet package in R29. A weighting procedure was included to up-weight hits, as described in Knight et al.30; in brief, the weighting has the effect of equalizing the number of hits and non-hits in the training set. Optimal parameters of lambda and alpha were selected for each elastic net model using 10-fold cross validation. Lambda is an overall penalty parameter. Alpha controls the proportion of weight assigned to both the sum of the absolute value of the coefficients and the sum of the squared value of the coefficients, which affects the degree of their sparsity. A range of combinations of lambda and alpha were investigated. The lambda and corresponding alpha that give a model a deviance one standard deviation above the model with the lowest deviance was selected.
Random forest is a collection of decision trees. The random forest models were implemented in Python using the scikit-learn package31. Two sets of random forest models were created, both using 10-fold cross validation. For the first set, we replicated Ritchie et al.’s random forest implementation by using scripts (e.g. gwava.py) provided on their online GWAVA FTP site (ftp://ftp.sanger.ac.uk/pub/resources/software/gwava/). For instance, bootstrap sampling was employed to form decision trees from bootstrap subset samples. To address the class imbalance in the datasets, non-hits were down-weighted through the balance_classes function created by Ritchie et al. and included in their random forest implementation. The balance_classes function selects a subset of non-hits that is equal to the number of hits in order to grow a tree. Furthermore, the subset of annotations used to determine the node split was set to the square root of the total number of annotations. This setting is the default setting for classification problems to determine the best split at each node of the decision tree32. Additionally, as done by Ritchie et al., we used 100 decision trees since we determined that the prediction scores and variable importance measures did not significantly differ past 100 trees.
Ritchie et al. used a minimum node size (min_samples_split) of 1. The minimum node size is the minimum number of samples required to split an internal node. We created another set of random forest models in which we adjusted the minimum node size. This parameter is dataset specific and a recommended setting is 10% of the total dataset32. Consider n to be the number of hits in the training dataset. For the second set of random forest models, we set the minimum node size to approximately 10% of 2n.
Support vector machine creates a hyperplane within a decision boundary space defined by support vectors to separate the classes in multidimensional space. The support vector machine models were implemented in Python through the scikit-learn package31. Kircher et al. did not use a weighting procedure as their training set was already balanced. To compare protocols in an unbiased manner, we used a subset of the training set in which we chose all hits and randomly selected an equal amount of non-hits. We performed a grid search using the tune function in order to determine the optimal cost parameter for a linear kernel. The cost parameter is a penalty (see chapter 9 in James et al.33 for details). Feature selection is critical to improving model performance and is intrinsically incorporated by the elastic net and random forest algorithms34. Feature selection must be implemented before using support vector machine, as there is no feature selection protocol built in. Kircher et al. utilized univariate logistic regression among other methods to select features that best predict genetic risk variants. In this paper our support vector machine models included those annotations that had a non-zero Beta coefficient from the corresponding elastic net models. We chose the annotations found to be important from elastic net, since this algorithm implements a more stringent feature selection protocol compared to random forest (see Results).
Assessment of model performance
We assessed model performance in the test set data by calculating the area under the receiver operating characteristic (ROC) curve using the R package ROCR35 (and verified using the R package pROC36). 95% confidence intervals were generated using 2000 bootstrap replicates also using pROC36. As another measure of model performance, we also examined the distribution of prediction scores assigned to the test set data with the aid of violin plots.
We investigated importance of the functional annotations through the Beta coefficient for elastic net. Similar to the output from a simple logistic regression, the larger coefficients are interpreted as more important to predicting genetic risk variants. For random forest we used Gini importance, which was also used in Ritchie et al. Gini importance is a scaled measure of Gini impurity averaged over all trees; it represents the improved capacity for correctly predicting variants that can be directly attributed to the annotation37. For support vector machine, feature weights can be obtained related to the construction of the hyperplane when a linear kernel is used38.
Performance for complex disease variants: Application to Schizophrenia GWAS
We tested the performance of the nine models based on the GWAS classifier in a schizophrenia GWAS context. We selected all sub-genome-wide-significant variants (5 × 10−8 < p < 1 × 10−6) from the first round of the GWAS by the Psychiatric Genomics Consortium (PGC1)24. For each of the nine models we obtained prediction scores for these variants and selected the variants from the first and fourth prediction score quartiles. For these variants we extracted the p-values from the larger second round of the GWAS (PGC2)2 and plotted these in quantile-quantile plots. Note that there is sample overlap in the discovery cohort (about 30%) of the smaller PGC1 in the larger PGC2. Sample details are provided as a Supplementary Table in the PGC2 paper2. We were able to determine for all models whether variants assigned higher scores were enriched in the variants with more significant p-values compared to variants with less significant p-values.
HGMD analysis
The nine models created by combinations of annotation sets and algorithms were assessed using two sets of the public release of the Human Gene Mutation Database (HGMD) variants provided to Ensembl in the fourth quarter of 2013 (provided by Graham Ritchie). In the first, we took all the variants (single nucleotide polymorphisms) in HGMD (N = 3,391) and chose non-hits/controls that fell within a kilobase of either side from the HGMD variant (for consistency with the way the controls were selected in Ritchie et al.12). Secondly, models based on the subset of non-exonic HGMD variants (N = 689) and non-exonic control variants were assessed. Additionally, the data was randomly split into 60% for training and 40% for testing. The same procedures for elastic net, random forest and support vector machine used in the GWAS Catalogue analysis were also conducted for the HGMD analyses.
Comparison of scores from the three papers: Application to Schizophrenia GWAS
In the effort for a more general comparison of the published methods as is, rather than looking specifically at the algorithm and annotations as done above, we additionally conducted the schizophrenia GWAS application using scores for the variants obtained directly from the published papers. This analysis is further described in Supplementary Text and the results are depicted in Supplementary Figure 4 and Supplementary Table 8.Computations were performed on either the CAMH Specialized Computing Cluster (SCC) or the General Purpose Cluster (GPC) supercomputer at the SciNet HPC Consortium [Ref. 39].
Additional Information
How to cite this article: Gagliano, S. A. et al. Smoking Gun or Circumstantial Evidence? Comparison of Statistical Learning Methods using Functional Annotations for Prioritizing Risk Variants. Sci. Rep. 5, 13373; doi: 10.1038/srep13373 (2015).
References
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Rivas, M. A. et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073 (2011).
Epi4K Consortium et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Disanto, G. et al. DNase hypersensitive sites and association with multiple sclerosis. Hum Mol Genet 23, 942–8 (2014).
Maurano, M. T. et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 337, 1190–1195 (2012).
Schaub, M. A., Boyle, A. P., Kundaje, A., Batzoglou, S. & Snyder, M. L inking disease associations with regulatory information in the human genome. Genome Res 22, 1748–59 (2012).
Gagliano, S. A., Barnes, M. R., Weale, M. E. & Knight, J. A Bayesian method to incorporate hundreds of functional characteristics with association evidence to improve variant prioritization. PLoS ONE 9, e98122 (2014).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Ritchie, G. R. S., Dunham, I., Zeggini, E. & Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 11, 294–296 (2014).
Hindorff, L. A., J. H., Hall, P. M., Mehta, J. P . & Manolio, T. A. A catalog of published genome-wide association studies. (2010). Available at www.genome.gov/gwastudies. Accessed: August 7, 2014.
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–5 (2014).
Stenson, P. D. et al. The Human Gene Mutation Database: 2008 update. Genome Med. 1, 13 (2009).
Parra, E., Eaton, K., Kavanagh, P., Edwards, M. & Krithika, S. Association study confirms that two OCA2 polymorphisms are involved in normal skin pigmentation variation in East Asian populations; (Abstract #1963S). Presented at the 64th Annual Meeting of The American Society of Human Genetics (October 19, 2014 in San Diego, CA).
Griswold, A. J. et al. Computational evaluation of the pathogenicity of noncoding sequence variants in autism spectrum disorder; (Abstract #1376T). Presented at the 64th Annual Meeting of The American Society of Human Genetics (October 21, 2014 in San Diego, CA).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinforma. Oxf. Engl. 26, 2069–2070 (2010).
The ENCODE Project Consortium. A User’s Guide to the Encyclopedia of DNA Elements (ENCODE). PLoS Biol 9, e1001046 (2011).
Pickrell, J. K. Joint Analysis of Functional Genomic Data and Genome-wide Association Studies of 18 Human Traits. Am J Hum Genet 94, 559–73 (2014).
Kichaev, G. et al. Integrating Functional Data to Prioritize Causal Variants in Statistical Fine-Mapping Studies. PLoS Genet. 10, (2014).
Strobl, C., Boulesteix, A. L., Zeileis, A. & Hothorn, T. Bias in random forest variable importance measures: illustrations, sources and a solution. BMC Bioinformatics 8, 25 (2007).
Boulesteix, A.-L., Janitza, S., Hapfelmeier, A., Van Steen, K. & Strobl, C. Letter to the Editor: On the term ‘interaction’ and related phrases in the literature on Random Forests. Brief. Bioinform. 16(2), 338–45 (2014).
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976 (2011).
Quang, D., Chen, Y. & Xie, X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinforma. Oxf. Engl. 31, 761–763 (2015).
Farh, K. K.-H. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Adzhubei I.A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249 (2010).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2013).
Knight, J., Barnes, M. R., Breen, G. & Weale, M. E. Using Functional Annotation for the Empirical Determination of Bayes Factors for Genome-Wide Association Study Analysis. PLoS ONE 6, e14808 (2011).
Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Malley, J. D., Kruppa, J., Dasgupta, A., Malley, K. G. & Ziegler, A. Probability machines: consistent probability estimation using nonparametric learning machines. Methods Inf Med 51, 74–81 (2012).
James, G., Witten, D. M., Hastie, T. & Tibshirani, R. in An introduction to statistical learning with applications in R Ch. 9, 337–372 (Springer: New York,, 2013).
Appavu, S., Rajaram, R., Nagammai, M., Priyanga, N. & Priyanka, S. in Advances in Computer Science and Information Technology (eds. Meghanathan, N., Kaushik, B. K. & Nagamalai, D. ) 501–511 (Springer: Berlin Heidelberg,, 2011).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011).
Hastie, T., Tibshirani, R. & Friedman, J. in The Elements of Statistical Learning: Data Mining, Inference and Prediction (Springer-Verlag: New York,, 2009).
Rosenbaum, L., Hinselmann, G., Jahn, A. & Zell, A. Interpreting linear support vector machine models with heat map molecule coloring. J. Cheminformatics 3, 11 (2011).
Loken, C. et al. SciNet: Lessons Learned from Building a Power-efficient Top-20 System and Data Centre. J. Phys. Conf. Ser. 256, 012026 (2010).
Acknowledgements
SciNet is funded by: the Canada Foundation for Innovation under the auspices of Compute Canada; the Government of Ontario; Ontario Research Fund—Research Excellence; and the University of Toronto. The CAMH SCC is funded by: The Canada Foundation for Innovation, Research Hospital Fund. We thank David Rotenberg and Jon Pipitone, SCC Administrators, for providing technical support. We thank Kathleen Askland, Nickhil Bhagwat and Malgorzata Maciukiewicz for support with the statistical learning. We thank Martin Kircher for support with CADD and for providing scripts to expand the 63 annotations to 949. We also thank Graham Ritchie for support with GWAVA. SAG is funded by the Peterborough K.M. Hunter Graduate Studentship and is also funded through the Armstrong Family via the CAMH Foundation. SAG is a fellow of CIHR STAGE (Strategic Training for Advanced Genetic Epidemiology)—CIHR Training Grant in Genetic Epidemiology and Statistical Genetics. This research was supported by funding from Training grant GET-101831. RR was funded through the Institute of Medical Science Summer Undergraduate Research Program. J.K. is currently funded by charitable donors at the CAMH Foundation: http://www.supportcamh.ca. J.K. holds the Joanne Murphy Professor in Behavioural Science. MEW is funded by the Higher Education Funding Council for England (HEFCE): http://www.hefce.ac.uk/. M.R.B. is funded by the National Institutes for Health Research (NIHR) and this work forms part of the portfolio of translational research of the NIHR Biomedical Research Unit at Barts.
Author information
Authors and Affiliations
Contributions
S.A.G. and R.R. conducted the analyses. S.A.G., R.R., M.R.B., M.E.W. and J.K. all helped to draft the manuscript and read and approved the final manuscript. All authors were involved in designing the experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gagliano, S., Ravji, R., Barnes, M. et al. Smoking Gun or Circumstantial Evidence? Comparison of Statistical Learning Methods using Functional Annotations for Prioritizing Risk Variants. Sci Rep 5, 13373 (2015). https://doi.org/10.1038/srep13373
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13373
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
