
Abstract
The cellular prion protein (PrPC) has been associated with a plethora of cellular functions ranging from cell cycle to neuroprotection. Mice lacking PrPC show an increased susceptibility to epileptic seizures; the protein, then, is neuroprotective. However, lack of experimental reproducibility has led to considering the possibility that other factors besides PrPC deletion, such as the genetic background of mice or the presence of so-called “Prnp flanking genes”, might contribute to the reported susceptibility. Here, we performed a comparative analysis of seizure-susceptibility using characterized Prnp+/+ and Prnp0/0 mice of B6129, B6.129, 129/Ola or FVB/N genetic backgrounds. Our study indicates that PrPC plays a role in neuroprotection in KA-treated cells and mice. For this function, PrPC should contain the aa32–93 region and needs to be linked to the membrane. In addition, some unidentified “Prnp-flanking genes” play a role parallel to PrPC in the KA-mediated responses in B6129 and B6.129 Prnp0/0 mice.
Similar content being viewed by others


Introduction
Although the role of the cellular form of the prion protein (PrPC) in living organisms has been intensively studied, a clear consensus concerning the physiological functions of this protein is still elusive and controversial. To date, existing evidence implicates PrPC in numerous distinct cellular processes, including cell proliferation and differentiation1,2, copper homeostasis3,4, oxidative stress5 and cell signaling6,7, among others.
The generation of different transgenic Prnp0/0 mice (mixed B6129 PrnpZrchl/Zrchl or co-isogenic 129/Ola PrnpEdbg/Edbg backgrounds) in the early 1990s did not reveal any relevant phenotypic alteration of the mutant mice8,9. However, subsequent studies identified an abundance of phenotypic alterations (e.g.,10), including depressive-like behaviour11, cognitive deficits12, age-dependent behavioural abnormalities13, altered olfaction14, peripheral myelin deficits15, altered circadian rhythms16 and an increased susceptibility to oxidative stress5 and glutamate excitotoxicity17,18,19. Indeed, different laboratories have described in these strains and in congenic B6.129 PrnpZrchl/Zrchl (B6129 mice backcrossed with C57BL/6 for several generations) an enhanced sensitivity to seizures after the administration of epileptogenic drugs such as kainic acid (KA), N-methyl-d-aspartic acid (NMDA), pilocarpine and pentylenetetrazol (PTZ), suggesting a neuroprotective role of the protein against excitotoxic insults (e.g.,17,18,19,20,21). However, some studies suggest that PrPC is not involved in KA-mediated excitotoxicity and that the observed differences are likely to be associated with the genetic background of the mice used in the experiments22,23,24. In fact, it was described some years ago how different mouse strains exhibit different vulnerability to the administration of glutamate and other seizure-inducing drugs, which correlates with differences in axonal sprouting and cell death in the hippocampal region (e.g.,25,26). For example, the hippocampus of the C57BL/6 background mouse, in contrast to FVB/N, is more resistant to the excitotoxic effects of KA25,27,28,29. Thus, these genetic differences dilute the specific participation of PrPC in KA-mediated cell death. However, these genomic influences do not per se explain the neuroprotective properties of PrPC observed in KA-treated neuroblastoma cell lines carrying different dosages of the Prnp gene (see below).
In addition to these genetic differences, a number of Prnp-flanking genes associated with the 129 genotype in B6129 PrnpZrchl/Zrchl mixed mice30 have recently been described. These genes are retained in Prnp0/0 progeny of congenic B6.129 PrnpZrchl/Zrchl after numerous (>10) crosses30,31 with C57BL/6 mice. SNP analysis of the backcrossed mice indicated that after 5 to 6 rounds the amount of C57BL/6-associated SNP increased from ≈60 up to ≈93%. Thus, B6.129 PrnpZrchl/Zrchl wild type and mutant mice may still differ at these additional polymorphic loci associated with Prnp32,33. One of these loci, the signal regulatory protein alpha (SIRPα) has recently been described as responsible for a previously PrPC-associated phagocytic function in macrophages33. More relevantly, overexpressing mice (Tg20) generated in mixed B6129 PrnpZrchl/Zrchl congenic mouse carried several copies of the polymorphic loci, since in most cases they are also crossed with previously backcrossed B6.129 PrnpZrchl/Zrchl mice30. In this scenario, it is reasonable to consider that the data obtained using Prnp0/0 or Tg20 (backcrossed or not with C57BL/6) in electrophysiological studies may yield conflicting results24,34,35,36. Indeed, lack of PrPC has been associated with long-term potentiation (LTP), GABA-mediated fast after hyperpolarization (AHP) and paired pulse facilitation (PPF) abnormalities in mutant mice (e.g.,37,38,39,40). However, regulatory participation of PrPC in neurotransmission and neuroprotection and in other cellular functions has also been shown with acute modulation of Prnp expression in neural cell lines and in other organisms (e.g., zebrafish) as models, to avoid the above-mentioned genetic problems of mice. For example, neuroblastoma (N2a) cells with reduced Prnp expression are more susceptible to KA in contrast to non-modified cells18,20. Functions of PrPC in neurotransmission are also reinforced since i) PrPC seems to be located in the synaptic cleft20,41,42,43; ii) several synaptic proteins (e.g., synaptophysin, synapsin Ib)44,45, glutamate receptor subunits (e.g., GluR6/7, NR2D, GluR1/2, mGluR1/5)19,20,46,47 and ion channels36 have been identified as interacting partners of PrPC; iii) PrPC regulates GluR6/7-mediated signalling through the modulation of the formation of the GluR6/7-PSD-95-MLK3 trimeric complex, which triggers the activation of the JNK3 apoptotic pathway in the hippocampus in response to KA administration20; and iv) it modulates the NMDA receptor48.
Due to these conflicting results, we aimed to clarify the participation of PrPC in KA-mediated excitotoxicity in vivo, using B6129 PrnpZrchl/Zrchl, B6.129 PrnpZrchl/Zrchl, 129/Ola PrnpEdbg/Edbg and FVB Prnp0/0 and Prnp+/+ mice and in vitro using N2a cells. In addition, we wished to explore whether the expression of truncated forms of PrPC (ΔF35 or ΔC4) lacking 32–134 and 32–93 residues of PrPC might increase the neurotoxic effects.
Our results indicate that FVB/N Prnp0/0 mice are not suitable for the in vivo model of KA-mediated excitotoxicity due to the intrinsic sensitivity to KA of FVB/N mice that hides clear differences49,50. More relevantly, both B6129 PrnpZrchl/Zrchl and co-isogenic 129/Ola PrnpEdbg/Edbg mice display more severe epileptic episodes and neurotoxic brain damage than their corresponding wild-type controls. Backcrossed B6.129 PrnpZrchl/Zrchl mice (with ≈93% of C57BL/6 SNPs) displayed lower numbers of seizures compared to B6129, indicating additional participation of Prnp-linked genes in KA-mediated effects in B6129 PrnpZrchl/Zrchl mice. However, our data using co-isogenic 129/Ola PrnpEdbg/Edbg (100% 129 genotype) and cell lines reinforce the notion that PrPC is neuroprotective in KA treatment. In addition, the expression of ΔF35 and ΔC4 in a B6129 PrnpZrchl/Zrchl background increased hippocampal cell death, especially in the CA3 region after KA injections. These in vivo data were also corroborated using N2a cells transiently expressing N-terminal truncated forms of PrPC. Lastly, our in vitro experiments also suggest that to be neuroprotective, PrPC should be bound to the plasma membrane by means of the GPI moiety.
Results
KA-induced seizures in several strains of wild-type and PrPC-null mice
A gene-targeting strategy to generate null mutations in mice is a powerful research tool to reveal in vivo the function of a single protein, as derived phenotypic alterations are usually attributed to the deleted gene. Nevertheless, behavioural alterations observed in null-mutant mice could result in some cases from the genetic background (see introduction, see also26,49,50). In order to determine a possible influence of non-Prnp genes in the susceptibility to KA-induced seizures, we compared the epileptic response in B6129 (n = 20), 129/Ola PrnpEdbg/Edbg (n = 9) and FVB/N Prnp0/0 (n = 7) and wild type mice (B6129, n = 16; 129/Ola PrnpEdbg/Edbg (n = 11); FVB/N (n = 8) (Fig. 1a and Supplementary Tables 1–6). A multiple administration protocol, consisting of three intraperitoneal injections of KA (10 mg/kg body weight) at 30 min intervals, was used in this experiment. Seizure intensity was analysed during the 4 hours after the first KA injection and scored as indicated in Methods. For an easier comparative analysis, data from grades I to IV were grouped together.

Comparison of KA-induced seizure profile in Prnp+/+and Prnp0/0 mice on B6129, B6.129, 129/Ola or FVB/N genetic backgrounds.
(a) Percentage of mice reaching seizure stages I to VI and KA-induced mortality. Adult animals were subjected to a multiple KA-injection protocol (10 mg/kg b.w.) and epileptic responses were analysed during 4 hours after the first KA injection (see Methods for details).(b) Example of the western blot detection of PrPC (6H4) expression in brain extracts obtained from B6129 PrnpZrchl/Zrchl Prnp+/+, B6129 PrnpZrchl/Zrchl Prnp0/0, 129/Ola PrnpEdbg/Edbg Prnp+/+, 129/Ola PrnpEdbg/Edbg Prnp0/0, FVB/N Prnp+/+ and FVB/N Prnp0/0 mice. Tubulin was used as a loading control. (c) Photomicrographs showing the pattern of neurodegeneration (Fluoro-Jade B staining) and DAPI staining of hippocampus in B6129 PrnpZrchl/Zrchl Prnp+/+, B6129 PrnpZrchl/Zrchl Prnp0/0, 129/Ola PrnpEdbg/Edbg Prnp+/+, 129/Ola PrnpEdbg/Edbg Prnp0/0, 24 hours after KA treatment. Dying cells are mainly located in the pyramidal cell layer of CA1 and CA3 regions of B6129 PrnpZrchl/Zrchl Prnp0/0 and 129/Ola PrnpEdbg/Edbg Prnp0/0 mice respectively. (d) Time course of stage VI seizure during the first 30 min, from 30–60 min and from 60–180 minutes, of adult B6129 PrnpZrchl/Zrchl Prnp0/0 vs B6.129 PrnpZrchl/Zrchl Prnp0/0. Data are presented as the mean ± S.E.M. of the number of seizures/number of animals. Note the lower levels of seizures in parallel with decreased 129 associated genes in B6.129 PrnpZrchl/Zrchl Prnp0/0 mice. Abbreviations: sr, stratum radiatum; sl, stratum lucidum; slm, stratum lacunosum-moleculare; sp, stratum pyramidale; so, stratum oriens. Scale bar represents 200 μm. Asterisks indicate statistical significance (***P < 0.01, Mann-Whitney U test).
After the behavioural study, mice were numbered and kept in separate boxes until histological studies. Percentages of the different strains of mice reaching each stage were represented (Fig. 1a). As indicated (Fig. 1a and Supplementary Tables 1–6), all mice achieved stages I–IV, developing hypoactivity and immobility shortly after the first injection. When comparing wild type vs Prnp-null mice, significant differences were observed in the percentage of mice reaching stages V and VI, with the B6129 PrnpZrchl/Zrchl and 129/Ola PrnpEdbg/Edbg much more susceptible to seizures than their respective Prnp+/+ controls (Fig. 1a and Supplementary Movies 1 and 2). In both groups, a high percentage of Prnp-null mice (100% and 75% respectively) developed loss of balance control and intermittent whole-body convulsions. More severe seizure activity, consisting of continuous seizures and/or ‘popcorn’ bouncing behaviour (stage VI) was also reported, leading to the death of 20% of the B6129 PrnpZrchl/Zrchl mice tested. In contrast, no B6129 Prnp+/+ or 129/Ola Prnp+/+ mice reached stage VI or died during experiments.
Distinct results were obtained when analysing FVB/N mice, since no significant differences were observed between FVB/N Prnp+/+ and FVB/N Prnp0/0 animals and only a 9% difference was observed between them (Fig. 1a and Supplementary Movie 3). FVB/N Prnp+/+ showed a high epileptic response at 10 mg/kg b.w of KA (≈50%), which was not observed in B6129 Prnp+/+ (≈30%) and 129/Ola Prnp+/+ (0%); it occurred in all described behavioural stages, but without any reported deaths. Although other unknown factors might be involved, such phenotypical differences between Prnp+/+ mouse strains could be related to small differences in PrPC expression levels, since our western blot analysis revealed lower PrPC expression in FVB/N Prnp+/+ (0.623 ± 0.04) compared to 129/Ola Prnp+/+ (0.92 ± 0.038) mice (t = 4.21, mean diff. = 0.30; 95% Cl of diff. = 0.66 to 0.54) but not between B6129 Prnp+/+ (0.84 ± 0.063) and FVB/N Prnp+/+ mice (t = 3,104, mean diff. = 0.22; 95% Cl of diff. = 0.−0.013 to 0.459), or between B6129 Prnp+/+ and 129/Ola Prnp+/+ (t = 1.112, mean diff. = −0.08; 95% Cl of diff. = −0.3166 to 0.1566) (Fig. 1b and Supplementary Fig. 1). It has been previously reported that excitotoxic damage derived from KA administration primarily affects pyramidal cells from CA1 and CA3 regions of the hippocampus. To corroborate that the increased cell death is associated with PrPC deletion, we focused on B6129 Prnp0/0 and 129/Ola Prnp0/0 mice. We obtained coronal brain sections (dorsal hippocampus) from these mice 24 hours after KA treatment and performed Fluoro-Jade B staining to determine the associated neurodegeneration in the hippocampus. Wild type mice were processed in parallel (Fig. 1c). B6129- Prnp0/0 mice displayed much more pronounced pyramidal cell damage, with higher numbers in CA1 than in CA3, than wild-type controls, in which no Fluoro-Jade B positive cells were observed. PrPC-dependent differences were also detected between 129/Ola- Prnp0/0 and 129/Ola-Prnp+/+ although cell death appeared to be restricted mostly to the CA3 region (Fig. 1c).
Taken together, these results support the notion that the genetic background of the different Prnp0/0 mice plays a role in KA susceptibility22,23, with the FVB/N background more prone to seizures than 129/Ola or the mixed genetic background B612926,49. The susceptibility of the FVB background49,50 probably masks participation of PrPC at this specific KA-dosage.
More relevantly, the present results demonstrate a role of PrPC in neuroprotection, since the phenotypic differences observed between Prnp0/0 and Prnp+/+ mice in B6129 PrnpZrchl/Zrchl (Supplementary Movie 1) are also reproduced in co-isogenic 129/Ola PrnpEdbg/Edbg mice (Supplementary Movie 2).
Decreased seizures in B6.129 PrnpZrchl/Zrchl compared to B6129 PrnpZrchl/Zrchl mice
In a second set of experiments we used B6.129 PrnpZrchl/Zrchl mice generated after crossing the original B6129 PrnpZrchl/Zrchl mice with C57BL/6 mice over several generations. In these experiments, B6129 PrnpZrchl/Zrchl mice were purchased directly from EMMA (Monterotondo, Italy) and they carried approximately ≈64% of C57BL/6 microsatellite markers (Charles River Laboratories) with ≈46% of non-C57BL/6 markers (129 in origin). We backcrossed these Prnp0/0 mice with C57BL/6 mice over several generations (8–10) to reduce the non-C57BL/6 microsatellite markers to ≈6.5–7%. The presence of C57BL/6 or 129 markers in all phenotypes used in the present study was determined by the genetic testing service at Charles River Laboratories. In the test, 110 microsatellite markers at approximately 15 cM intervals were analyzed, spreading across the 19 autosomes and the X chromosome, which distinguishes among 129 microsatellite markers ranging from 92 to 94% of C57BL/6 (in B6.129 PrnpZrchl/Zrchl mice). Thus B6129 and B6.129 PrnpZrchl/Zrchl mice were treated with 8 mg/kg b.w. (B6129 n = 8 and B6.129 PrnpZrchl/Zrchl n = 6) or 10 mg/kg b.w (B6129 n = 8 and B6.129 PrnpZrchl/Zrchl n = 9) of KA and the number of seizures was determined during the first 30 minutes, from 30 to 60 minutes and from 60 to 180 minutes postinjection. In this experiment each mouse received a KA injection every 30 minutes as above. Data revealed a relevant number of seizures in B6129 PrnpZrchl/Zrchl Prnp0/0 mice compared with B6.129 PrnpZrchl/Zrchl Prnp0/0 (KA 8 mg/kg P = 0.0021; KA 10 mg/kg P = 0.0086; Mann-Whitney U test confidence interval (CI) = 95% (Fig. 1d and Supplementary Movie 1). In addition, the differences between the numbers of seizures can be observed in both KA concentrations (8 and 10 mg/kg b.w). These data indicate that although PrPC plays a role in the increased susceptibility to KA as demonstrated above, the 129 associated genes also play a role in the observed results.
Increased susceptibility in B6.129-Prnp0/0 and 129/Ola- Prnp0/0 correlates with enhanced cell death, inflammatory markers and astrogliosis
Parallel histological sections to those processed in Fig. 1 KA-induced astrocytic activation and inflammatory response in B6129-Prnp0/0 (n = 5), 129/Ola- Prnp0/0 (n = 5) mice at their respective controls (n = 5 each genotype) (Fig. 2a). Immunohistochemical analysis of GFAP-positive cells in the hippocampus of PrPC-null mice (either B6129 or 129/Ola) revealed large numbers of positive cells in the hippocampal CA1-3 when compared to Prnp+/+ controls (B6129-Prnp0/0 75.7 ± 1.24 vs B6129-Prnp+/+ 43.4 ± 1.06; P = 0.005 Mann-Whitney U test. 129/Ola- Prnp0/0 53.7 ± 1.44 vs 129/Ola-Prnp+/+ 42.9 ± 0.92; P = 0.0047 Mann-Whitney U test. Immunoreactive cells showed hypertrophic cell bodies and thicker glial processes (Fig. 2a). Moreover, quantitative real-time PCR data obtained from hippocampal samples 6 hours after KA-administration showed upregulation of the main pro-inflammatory markers TNFα (4.7-fold increase in B6129-Prnp0/0 (n = 3) P = 0.0046, Mann-Whitney U test and 3.8-fold increase in 129/Ola-Prnp0/0 (n = 3) (P = 0.0008, Mann-Whitney U test) vs respective wild type (B6129 n = 3 and 129/Ola n = 3) and IL1β (3.7-fold increase in B6.129- Prnp0/0 and 3.0-fold increase in 129/Ola- Prnp0/0 vs respective wild type (B6.129- Prnp0/0 P = 0.0046 and 129/Ola Prnp0/0 P = 0.0004 and Mann-Whitney U test)) (Fig. 2b).

Increased astrogliosis, TNFα and IL1β expression in the hippocampus of B6129 PrnpZrchl/Zrchl Prnp0/0 and 129/Ola PrnpEdbg/Edbg Prnp0/0.
(a) Examples of GFAP-positive immunoreactive cells in the stratum radiatum of the hippocampal CA1-3, 24 hours after KA administration in hippocampal coronal sections of B6129 PrnpZrchl/Zrchl Prnp+/+, B6129 PrnpZrchl/Zrchl Prnp0/0, 129/Ola PrnpEdbg/Edbg Prnp+/+, 129/Ola PrnpEdbg/Edbg Prnp0/0. Both Prnp-null mice show greater numbers of reactive astroglia than their wild-type controls. (b) RT-qPCR of TNFα and IL1β mRNA levels from hippocampal RNA extracts obtained from B6129 PrnpZrchl/Zrchl Prnp+/+, B6129 PrnpZrchl/Zrchl Prnp0/0, 129/Ola PrnpEdbg/Edbg Prnp+/+, 129/Ola PrnpEdbg/Edbg Prnp0/0 mice 6 hours after the last KA treatment (at 60 minutes). Plotted data (mean ± S.E.M.) were obtained from three independent experiments and represented as mean fold change induction. Abbreviations are as in Fig. 1. Scale bars represent 100 μm. Asterisks indicate statistical significance (***P < 0.01 Mann-Whitney U test).
Chemical elimination of the GPI domain and of PrPC reduces neuroprotection to KA in Prnp-transfected N2a cells
The GPI anchor in PrPC, as in other GPI-proteins, is not only necessary for the stability and attachment of the protein to the cell surface, but also for its association to specialized membrane microdomains (e.g., lipid rafts), its intracellular traffic and signal transduction events. In addition, the GPI group has been suggested as playing a role in prion disease toxicity, as transgenic mice expressing secreted forms of PrPC lacking its GPI-moiety showed no clinical symptoms despite accumulating PrPSc in plaques51. Because in our microsatellite analysis the C57BL/10-PrPGPIless mice (Tg44+/+ kindly provided by Dr. B. Chesebro) contained non-B10 regions in chromosome 3 and in chromosome 2 flanking the Prnp locus, we decided to check whether the degradation of the GPI binding domain of the PrPC leading to a decreased PrPC in the plasma membrane could overcome the neuroprotective function of PrPC (Fig. 3a–c). In addition, this approach would determine whether the neuroprotective effect against KA treatment of PrPC takes place at the membrane or intracellularly. Thus, N2a cells were treated either with Phospholipase C (PLC) enzyme or Glimepiride, a sulphonylurea approved for the treatment of diabetes mellitus, inducing the release of PrPC from the surface of prion-infected neuronal cells, which releases PrPC from the surface of neuronal cells52 (see methods for details). Decreased levels of PrPC after treatments were detected with western blot (pcDNA = 0.39 ± 0.021; pcDNA-PrPC = 0.91 ± 0.025; pcDNA-PrPC + PLC = 0.45 ± 0.013; pcDNA-PrPC + Gli = 0.496 ± 0.014) (Fig. 3a and Supplementary Fig. 2) or with immunocytochemistry in non-permeabilized cells (Fig. 3b) (pcDNA CTCF = 1.63 ± 0.09; pcDNA-PrPC CTCF = 6.44 ± 0.12. pcDNA-PrPC + Gli CTCF = 2.82 ± 0.20; pcDNA-PrPC + PLC CTCF = 2.063 ± 0.15. pcDNA vs pcDNA-PrPC t = 7.40, mean diff. = −4.8, 95% CI of diff. = −6.88 to −2.72. pcDNA-PrPC vs pcDNA-PrPC + Gli t = 5.31, mean diff. = 3.6, 95% CI of diff. = 1.43 to 5.78. pcDNA-PrPC vs pcDNA-PrPC + PLC t = 6.02, mean diff. = 4.37, 95% CI of diff. = 2.04 to 6.70. WST-derived results indicate that both treatments increased the sensitivity to KA (pcDNA vs pcDNA-PrPC t = 4.38, mean diff. = −0.27, 95% CI of diff. = −0.44 to −0.10. pcDNA-PrPC vs pcDNA-PrPC + PLC t = 2.789, mean diff. = −0.175, 95% CI of diff. = −0.34 to −0.005. pcDNA-PrPC vs pcDNA-PrPC + Gli t = 4.46, mean diff. = 0.28, 95% CI of diff. = 0.11 to 0.45 (Fig. 3c).

Chemical release of PrPC from the cell surface impairs the neuroprotective function in KA-treated N2a cells.
(a) Example of western blot determination of PrPC levels in protein extracts of pcDNA, pcDNA-PrPC, pcDNA-PrPC + PLC and pcDNA-PrPC + Gli treated N2a cells. (b) Microphotographs illustrating PrPC levels in cells transfected with pcDNA or pcDNA-PrPC and treated with PLC (pcDNA-PrPC + PLC) or Gli (pcDNA-PrPC + Gli). PrPC (green) was detected in non-permeabilized cells with 6H4 antibody. Note the decrease in PrPC after PLC and Gli treatments in (a) and (b). (c) Histogram showing the decrease in surviving cells after KA-treatment (5 mM overnight) determined with the WST-1 viability test after PLC or Gli treatment of pcDNA-PrPC transfected cells. Data (mean ± S.E.M.) were obtained from three independent experiments and represented as mean fold change. Scale bars represent 100 μm. Asterisks indicate statistical significance (**P < 0.05, Bonferroni post hoc test).
Enhanced cell death in the CA3 hippocampal region ΔF35 and ΔC4 mice compared to B6129 mice
Prnp0/0 ΔF35 (ΔF35) and Prnp0/0 ΔC4 (ΔC4) mice (Fig. 4a) were generated some years ago by nuclear injections of constructs into fertilized oocytes from B6129 PrnpZrchl/Zrchl mice53. The number of copies of the construct was estimated as 25 for ΔC4 and 70 for ΔF35 mice. However, brain extracts obtained from these mice revealed similar levels of PrP−ΔC4 (Prnp+/+ = 0.755 ± 0.075; PrP−ΔC4 = 0.81 ± 0.03; P = 0.4; CI = 95%, Mann-Whitney U test) to wild-type but lesser amounts of PrP−ΔF35 than wild-type (Prnp+/+ = 0.91 ± 0.01; PrP−ΔF35 = 0,70 ± 0.021; P = 0.0048, CI = 95%, Mann-Whitney U test) (Fig. 4b and Supplementary Fig. 3). Nevertheless only ΔF35 mice showed cerebellar degeneration at around 60 days of life53 (Supplementary Fig. 4). Irrespective of the cell type, the expression of the truncated form may induce per se cell death in vivo54 as well as in vitro55. Thus in the next experiments we treated these mice with 8 mg/kg b.w. of KA following the above-mentioned protocol. Behavioural results reported a similar evolution between ΔC4 (n = 11) and ΔF35 (n = 15) mice compared to B6129 PrnpZrchl/Zrchl Prnp0/0 (n = 35) and B6129 Prnp+/+ (n = 30) (Fig. 4c and Supplementary Movie 4). However, the number of dead was greater in ΔC4 (≈38%) and ΔF35 (≈15%) compared to B6129 PrnpZrchl/Zrchl Prnp0/0 mice. These results also correlate with an increased presence of Fluoro-Jade B cells observed in histological sections of the hippocampus, especially in the CA3 region (ΔF35 = 87.12 ± 0.41; ΔC4 = 104.62 ± 0.088; B6129 PrnpZrchl/Zrchl = 13,37 ± 0.055. B6129 PrnpZrchl/Zrchl vs ΔF35 t = 7.577; mean diff. = −73.75, 99% Cl of diff. = −115.4 to −32.1. B6129 PrnpZrchl/Zrchl vs ΔC4 t = 9.375; mean diff. = −91.25, 99% CI of diff. = −132.9 to −49.6, Bonferroni post hoc test). (Fig. 4d). Taken together, these results indicate that the expression of the truncated form including the octarepeat region (OR) with or without the Central Domain (CD) of PrPC potentiates the effects of the KA (<10 mg/kg b.w.) and they highlight the octarepeat domain as a key candidate in the neuroprotective functions of PrPC.

KA-induced seizures and neurotoxicity in ∆C4 and ∆F35 mice.
(a) Scheme of PrPC and N-terminal truncated forms overexpressed in B6129 PrnpZrchl/Zrchl Prnp0/0 ∆C4 and B6129 PrnpZrchl/Zrchl Prnp0/0 ∆F35 transgenic mice. (b) Example of western blot detection of PrPC (6H4) expression in brain extracts obtained from untreated B6129 PrnpZrchl/Zrchl Prnp+/+and B6129 PrnpZrchl/Zrchl Prnp0/0, B6129 PrnpZrchl/Zrchl Prnp0/0 ∆C4 and B6129 PrnpZrchl/Zrchl Prnp0/0 ∆F35 mice. Tubulin was used as a loading control. (c) KA-induced seizure sensitivity in adult B6129 PrnpZrchl/Zrchl Prnp0/0 ∆C4 mice in comparison to B6129 PrnpZrchl/Zrchl Prnp+/+ and B6129 PrnpZrchl/Zrchl Prnp0/0 (upper panel). KA-induced seizure sensitivity in 5–7 week-old B6129 PrnpZrchl/Zrchl Prnp0/0 ∆F35 mice in comparison to B6129 PrnpZrchl/Zrchl Prnp+/+ and B6129 PrnpZrchl/Zrchl Prnp0/0 (lower panel). Bars represent the percentage of mice reaching each stage. (d) Fluoro-Jade B staining in hippocampal sections of B6129 PrnpZrchl/Zrchl Prnp+/+, B6129 PrnpZrchl/Zrchl Prnp0/0, B6129 PrnpZrchl/Zrchl Prnp0/0 ∆C4 and B6129 PrnpZrchl/Zrchl Prnp0/0 ∆F35 mice 24 hours after KA treatment (8 mg/kg b.w.). The quantification of the Fluoro-Jade B-positive cells in the CA3 region is shown in the right plot. Scale bars represent 200 μm. Abbreviations: DG, Dentate gyrus; CA1-3, hippocampal regions 1 and 3; h, hilus; gl, granule cell layer; ml, molecular layer; sr, stratum radiatum; slm, stratum lacunosum-moleculare; sl, stratum lucidum; sp, stratum pyramidale; so, stratum oriens. Asterisks indicate statistical significance (***P < 0.01, Bonferroni post hoc test).
Acute transfection of pcDNA-PrP∆CD but not pcDNA-PrP∆F35 protects N2a cells from KA excitotoxicity in vitro
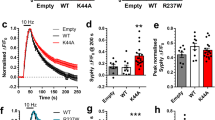
In order to corroborate in vitro the participation of the OR in PrPC-mediated neuroprotection to KA, we performed a viability assay using N2a cells. Cells were transfected with vectors encoding either the full length of Prnp or two truncated forms lacking CD (PrP∆CD, residues 95–133), which bridge the flexible amino proximal tail and the globular carboxy proximal domain, or else a longer deletion including the central domain (CD) plus the OR (PrP∆F35, residues 32–134) (Fig. 5a). After transfection, levels of PrPC and its truncated forms in transfected cells were checked with western blotting (Fig. 5b and Supplementary Fig. 5). In addition, the expression of the truncated forms was strictly modulated to avoid inducing cell death during the experiments. Transfected cells were treated overnight with 5 mM KA and further processed to WST-1 assays. Colorimetric WST-1 assay showed that PrPC and PrP∆CD transfection increased cell culture viability after KA treatment, an effect that was not observed in PrP∆F35 transfected cells (pcDNA = 0.54 ± 0.005; pcDNA-PrPC = 0.94 ± 0.06; pcDNA-PrP∆CD = 1.047 ± 0.012 and pcDNA-PrP∆F35 = 0.88 ± 0.03. pcDNA vs pcDNA-PrPC t = 36.18; mean diff. = −0.4, 99% CI of diff. = −0.45 to 0.34. pcDNA vs pcDNA-PrP∆CD t = 45.83; mean diff. = −0.506, 99% CI of diff. = −0.55 to 0.45. pcDNA-PrP∆CD vs pcDNA-PrP∆F35 t = 15.08, mean diff. = 0.166, 95% CI of diff. = 0.1282 to 0.20). pcDNA-PrPC vs pcDNA-PrP∆F35 t = 5.42, mean diff. = 0.06, 95% CI of diff. = 0.021 to 0.09) (Fig. 5c). These results reinforce the idea of the OR domain participating in the described neuroprotective role of PrPC to excitotoxic damage in N2a cells.

KA-excitotoxicity in N2a cells transiently transfected with PrPC or PrP-∆CD and PrP-∆F35 constructs.
(a) Scheme illustrating domain organization in PrPC and its related PrP∆CD and PrP∆F35 mutants. (b) Example of western blot determination of the different PrPC constructs employed in the experiment. (c) WST-1 viability assay performed over N2a cells previously transfected with pcDNA3.1 (empty vector, pcDNA), pcDNA-PrPC, pcDNA-PrP∆CD and pcDNA-PrP∆F35 and treated overnight with 5 mM KA. Data were normalized with untreated controls. A clear reduction in cell viability was observed in pcDNA-PrP∆F35 transfected cells when compared to those overexpressing pcDNA-PrPC. In contrast, pcDNA-PrP∆CD transfected cells showed similar viability to pcDNA-PrPC transfected cells. Histograms represent the mean ± S.E.M. of three different experiments. Asterisks indicate statistical significance (**P < 0.05, ***P < 0.01, Bonferroni post hoc test).
Discussion
Role of PrPC in neuroprotection against KA
The diversity of phenotypic changes described in Prnp-knockout mice has hindered the study of the physiological function of PrPC10. In fact the reported differences between Prnp+/+ and Prnp0/0 mice have led to certain controversial results, especially in terms of electrophysiology and susceptibility to excytotoxic insults. Several pieces of evidence, including some from previous studies by our group using B6.129 PrnpZrchl/Zrchl 2,18,21,40, support the idea that Prnp-knockout mice are more susceptible to KA, NMDA and PTZ, exhibiting an enhanced epileptic response and neurotoxicity in the hippocampus when compared to wild type controls17,19. Contradictory results have been published by other groups, who described an elevated threshold for epileptiform activity in Prnp0/0 hippocampal slices exposed to bicuculline, PTZ or zero-magnesium conditions24. Similar discrepancies have been found when analysing neurotransmission-associated parameters in mice devoid of PrPC34,35,37,38. Due to this lack of reproducibility between groups, the possibility that reported phenotypes could be attributed to external factors, such as different mice strains (e.g.,25 or 32) or experimental procedures (age of mice, KA concentration, treatments, etc.) must be strongly considered. Indeed, age-dependent loss of LTP in PrPC-null mice56,57 has also been identified.
More recently, a comparative analysis between Prnp-knockout mouse strains has led to the proposal that the increased sensitivity to KA-induces seizures in Prnp-knockout mice is not associated with the absence of the protein but rather with inappropriate comparison with wild-type controls that differ in genetic background22,23,58. In this study, background influence in susceptibility to excitotoxic damage was corroborated by evaluating the onset of epileptic seizure of wild-type mice from B6129, B6.129,129/Ola and FVB/N strains. Our results show that FVB/N wild-type mice showed more seizures than B6129 and 129/Ola mice, corroborating previous studies25,26,59. In addition, 129/Ola wild-type showed increased expression of PrPC that may also reduce the number of grade VI seizures.
In our study we observed the relevant effects of intraperitoneal injection of KA in B6129 PrnpZrchl/Zrchl Prnp0/0 (with ≈46% 129 microsatellites), B6.129 PrnpZrchl/Zrchl Prnp0/0 (with ≈6–7% 129 microsatellites) and 129/Ola-Prnp0/0 mice (with 100% 129 background) compared with the appropriate wild-type controls. Results indicate a decline in the epileptic seizures when low numbers of 129 microsatellites are present in B6.129 PrnpZrchl/Zrchl Prnp0/0. These results point to polymorphisms of some of the “Prnp- flanking genes” as masking the participation of PrPC in neuroprotection, as reported in other processes30. Analysis of the 9 genes functions using gene ontology and Pubmed searches indicates that Prex160, SIRPα61, Traf162, Thbs163,64, Rmdn365, Tyro366,67, Slc30a468, Mertk69 and B2m70 are involved in neurotransmission, LTP and neuroprotection. Whether polymorphisms in these genes participate in these effects warrants further study. However, the phenotypic differences observed between 129/Ola Prnp+/+ and 129/Ola Prnp0/0 mice could only be associated with Prnp-deletion, as no possible genetic variability exists between the two. These results differ from those recently published by Striebel and coworkers23 who didn’t observe PrPC-linked neuroprotection despite using the same mouse strain. These contradictory results may be due to KA-dosage or to minor differences in the experimental procedure. Furthermore, we must not forget that this neuroprotective property of PrPC is further supported by other in vivo models, such as transient knockdown of Prnp homologs in zebrafish71 and also some in vitro experimental approaches including PrPC downregulation and detachment of the plasma membrane in neuronal primary cultures and neuroblastoma cell lines (18,19,20 and present results).
In conclusion, our findings support the notion that PrPC is involved in neuroprotection to KA-induced seizures and excitotoxicity and that it actively participates in the increased epileptic response observed in mice devoid of PrPC. In parallel, as yet unknown factors associated with Prnp-flanking genes also affect KA susceptibility.
The neuroprotective function of PrPC depends on membrane anchoring
Our results indicate that the neuroprotective function of PrPC against KA in N2a cells depends on membrane anchoring. The interaction of different regions of PrPC with plasma membrane through the GPI domain has been considered necessary to induce the clinical symptoms in GPI-negative transgenic mice (C57BL/10-PrPGPIless mice)51,72. In fact, the injection of antibodies directed to the α1 and α3 regions of the PrPC induces neurotoxic effects73. These results have also been corroborated in newly developed mice lacking the globular domain (FTgpi mice)74. In fact, these results corroborate previous observations75. Taken together, these studies suggest that the proximity of the flexible tail (N-terminal domain) to the plasma membrane triggers intracellular oxidative stress responses leading to cell death74,76. Under this scenario we can consider that our data reinforce the idea that the integrity of the N-terminal domain is mandatory for neuroprotection (see below) as well as the notion that membrane interaction is a necessary part of the neuroprotective function reported in vivo.
The neuroprotective function of PrPC is abolished in the absence of the OR of PrPC
Structural analysis of PrPC architecture has determined different functionally relevant domains in this protein (see77,78 for review). Besides the highly conserved hydrophobic domain, the flexible and unstructured N-terminus region includes a copper binding site consisting of four tandem repeats of the sequence PHGGGWGQ, which seems to be involved in the endocytic process of the protein79 and copper homeostasis3. In fact a recent study indicates a relevant role of copper binding in the neuroprotective function of PrPC modulating NMDA receptor48. In our study we developed in vivo as well as in vitro tests to check the involvement of the OR domain as a mediator of PrPC neuroprotective function in a model of KA treatment. The behavioural and histological data presented here by ΔC4 and ΔF35 mice revealed increased cell death in the hippocampus of the KA injected mice compared to B6129 Prnp0/0 (genetic background of these mice). In parallel, the CD is not involved in the neuroprotective functions of PrPC (at least in N2a cells) since their absence does not modify these properties when compared to full length PrPC, in contrast to the absence of the OR regions.
The overexpression of PrP−ΔF35 in a B6129 PrnpZrchl/Zrchl Prnp0/0 background leads to cerebellar neurodegeneration53, but not hippocampal degeneration55 (Supplementary Fig. 4). In contrast, ΔC4 mice (with similar background) do not show cerebellar or hippocampal degeneration, but when subjected to controlled ischemia show significantly greater oxidative stress damage when compared to wild type mice80. This also happens when PrP−ΔF35 is overexpressed in HEK293 cells, leading to increased Caspase 3 activity in transfected cells and cell death55. Truncated forms of PrPC lacking the OR interfere with PrPC endocytosis via clathrin-coated vesicles and beta-cleavage of PrPC, respectively, thereby impairing the antioxidative functions of PrPC81,82. Thus it is reasonable to consider that cells with an intrinsic deficit in oxidative stress homeostasis may also be more prone to KA treatment if the appropriate KA receptors are also expressed, as happens in the hippocampus83,84,85.
As indicated above, using antibody-mediated degeneration, Sonati et al., demonstrated that ligands directed to the α1 and α3 helices of the PrPC globular domain induce cerebellar cell death by activating oxidative stress that can be overcome by deletions in the OR region73. This also happens in FTgpi mice lacking the α1–α3 helix region of the PrPC 74. In addition, mice lacking the ΔCD also reported white matter pathology and peripheral neuropathy86. Surprisingly, the reported degeneration of the ΔCD mice could be reversed by coexpression of PrPC lacking all octarepeats86. These data are in contrast to a recent study indicating that the antibody ICSM18 (recognizing aa143–153 of PrPC) does not induce cell death87. In our experiments, we observed that cells transfected with PrP−ΔCD are able to overcome KA-mediated cell death as are those transfected with full length PrPC, in contrast to PrP−ΔF35 transfected N2a cells. Our in vitro experiments are different from the in vivo situation, since an effect of ligands in the CD regions is unlikely; rather, they suggest a parallel effect of KA excytotoxicity plus the homeostatic imbalance induced by the absence of the 32–93 region of the overexpressed PrPC. Despite the existing data, the precise mechanism underlying OR-dependent neuroprotection remains to be elucidated in the above-mentioned studies73,87,88.
In conclusion, our study dissects the effects of the intraperitoneal injection of various doses of KA in several Prnp mouse models and indicates that: i) PrPC plays a role in neuroprotection in KA-treated cells and mice; ii) for this role PrPC should be linked to the membrane; iii) polymorphisms of some unidentified “Prnp-flanking genes” play a parallel role to PrPC in the KA-mediated responses in B6129 and B6.129 PrnpZrchl/Zrchl Prnp0/0 mice; and iv) the absence of the aa32–93 region negatively affects the neuroprotective function of PrPC in KA-treated cells.
Methods
Reagents and antibodies
KA, glimepiride and phospholipase C were purchased from Sigma (Poole Dorset, UK). Fluoro-Jade B was from Millipore (Billerica, MA), SYBR green (Applied Biosystems, USA) and WST−1 reagents were from Roche (Basel, Switzerland). Lipofectamine plus was from Invitrogen (Carlsbad, CA). The following antibodies were used in this study: Anti-PrP SAF61 mouse monoclonal antibody (1:1000 diluted) antibody was from Spi-Bio (Cayman Chemical, Massy, France) and anti-PrP 6H4 mouse monoclonal antibody (1:5000 diluted western blotting and 1:250 immunocytochemistry) antibody was from Prionics (Schlieren, Switzerland). Mouse monoclonal antibody anti-tubulin (1:10000 diluted) was from Sigma (Poole Dorset, UK) and rabbit-raised polyclonal antibody against GFAP (1:500 diluted) was from Millipore (Billerica, MA).
Animals
Adult male C57Bl6/129sv-Prnp0/0 (B6129 PrnpZrchl/Zrchl Zurich I) mice were purchased from the European Mouse Mutant Archive (EMMA, Monterotondo, Italy). The FVB/N animals were either wild-type or homozygous for the deletion of Prnp gene. 129/Ola PrnpEdbg/Edbg Prnp+/+ and 129/Ola Prnp0/0 9 were obtained from Dr. J. Manson (Edinburgh). B6129 PrnpZrchl/Zrchl mice were backcrossed for 8 generations to obtain 6–7% of 129 microsatellites (B6.129 PrnpZrchl/Zrchl). Transgenic B6129 Prnp0/0 PrP∆C4 89 and B6129 Prnp0/0 PrP∆F35 53 were obtained from Prof. A. Aguzzi. To avoid putative background-specific differences between mice, all of the experiments were conducted using littermates derived from heterozygous (Prnp+/0 ΔF35+) and Prnp0/0 parents. Specific primers for Prnp genotyping were designed in our laboratory based on the original P10 and P3 primers described elsewhere8. PrP∆F35 transgene was detected by using 5′-CCTGGGACTCCTTCTGGTACCGGGTGACGC-3′ and 5′-CAACCGAGCTGAAGCATTCTGCCT-3′ set of primers and PrP∆C4 with 5′-GGCTGGGCTTGTTCCACTGATTATGGG-3′ and 5′-CAACCGAGCTGAAGCATTCTGCCT-3′. Tg44+/+ mice expressing anchorless PrPC 51 on a C57Bl/10 background were obtained from Dr. Bruce Chesebro (Laboratory of Persistent Viral Diseases, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, Hamilton, MT 59840, USA). A total of 83 litters (193 animals) were used in the present study. All experiments were performed under the guidelines and protocols of the Ethical Committee for Animal Experimentation (CEEA) of the University of Barcelona and the protocol for the use of animals in this study was reviewed and approved by the CEEA of the University of Barcelona (CEEA approval #115/11 and 141/15).
KA administration in mice and seizure analysis
Convulsive non-lethal seizures in mice were induced by administration of KA in a multiple dose protocol. Fresh KA solution was prepared for each experiment. Animals were weighed and intraperitoneally injected with 8 or 10 mg/kg KA (b.w.) dissolved in 0.1 M PBS, pH 7.2. at 0 min, 30 min and 60 min. In parallel mice, 0.1 M PBS, pH 7.2 was injected as control (vehicle). Adult (2–3 months old) animals were used in all experiments except for B6129 PrnpZrchl/Zrchl Prnp0/0 PrP∆C4 PrP∆F35 and their respective controls, which were used at 5–7 weeks-old due to their described early lethality53.
After KA-injection mice were distributed in boxes (1–5 mouse/box) and the behaviour of the mice was recorded for 4 hours using a digital video camera (SONY DCR-HC30E Digital video camera). Seizure intensity was evaluated during the 4 hours after the first injection using the following criteria: grades I-II: hypoactivity and immobility, grades III-IV: hyperactivity and scratching, grade V: loss of balance control and intermittent whole-body convulsions and grade VI: continuous seizures and bouncing activity (commonly referred to as ‘popcorn’ behaviour). The characterization of seizure intensity was developed in two stages. First seizures were analysed in situ after KA-injections in the Molecular and Cellular Neurobiotechnology Laboratory (IBEC) by P.C., F.Ll. and A.M-A. (B6129 PrnpZrchl/Zrchl Zurich I, B6.129 PrnpZrchl/Zrchl, B6129 Prnp0/0 PrP∆C4, B6129 Prnp0/0 PrP∆F35, FVB/N Prnp0/0 mice strains and its respective controls) and in the Centro de Investigación en Sanidad Animal (CISA-INIA) by P.C, P.A-C., J.C.E. (129/Ola Prnp0/0 and 129/Ola PrnpEdbg/Edbg Prnp+/+). After this initial characterization that included time of the appearance of seizures, scale of seizures, etc, video recordings of each experiment were analysed by J.A.D.R. at IBEC and J.M.T. at CISA-INIA without knowledge of the results of the previous in situ analysis. Afterwards, a contingency table was generated by comparing both analyses and the final data were plotted. Grades I to IV were grouped together for better data representation, as all animals tested reached these four epileptic stages. The statistical analysis of the obtained data was performed using Mann–Whitney U non-parametric test using Prism 5.0c (Mac OsX, Grahpad). Data are presented as percentage or as mean ± standard error of the mean (S.E.M.). A value of ***P < 0.01 was considered statistically significant.
Western blotting
Brain samples from non-treated mice were homogenized (10% wt/vol) in ice-cold lysis buffer—50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% (wt/vol) Triton X-100, 0.5% (wt/vol) Nonidet P-40 (IGEPAL; Sigma), glycerol 10%, 1 mM EDTA, 1 mM EGTA and protease and phosphatase inhibitors—using a motor-driven, glass-Teflon homogenizer in ice and then centrifuged at 15,000g for 20 min. Protein concentration was quantified with BCA kit (Pierce). Protein extracts were boiled in Laemmli sample buffer at 100 °C for 5 min. Equal amounts of total protein were separated with 6–10% SDS-PAGE electrophoresis, electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore) and probed with indicated antibodies. Visualization of bound antibodies was performed using goat anti-mouse HRP (1:4000 diluted; Dako, Glostrup, Denmark) and the ECL Plus kit (Amersham-Pharmacia Biotech, GE Healthcare Bio-Sciences, Piscataway, NJ, USA).
Densitometry and statistical processing of processed films
For quantification, developed films were scanned at 2400 × 2400 dpi (i800 MICROTEK high quality film scanner) and the densitometric analysis of the different PrPC bands was performed in each case using Quantity One Image Software Analysis (Biorad). Each densitometric value of PrPC and truncated form ΔC4, ΔF35 and ΔCD (0–255 gray scales) was normalized with the corresponding Tubulin densitometric values (0–255 gray scale). Three different experiments were used in each analysis unless specified. The statistical analysis of the obtained data was performed using Bonferroni post hoc test (Multiple comparison test) or Mann-Whitney U non-parametric test using Prism 5.0c (Mac OsX, Grahpad). Data are presented as mean ± standard error of the mean (S.E.M.). A value of **P < 0.05 was considered statistically significant.
Fluoro-Jade B staining
Mice were perfused with 4% paraformaldehyde dissolved in 0.1 phosphate buffer, pH 7.3 24 hours after the first KA injection, post-fixed overnight in the same fixative and cryoprotected in 30% sucrose. 30 μm-thick coronal brain sections were obtained in a freezing microtome (Leica, Wetzlar, Germany). Sections containing dorsal hippocampus (Bregma = −1.2 to −1.990) were rinsed for 2 h in 0.1 M Tris, pH 7.4, mounted and air dried at room temperature overnight. The next day, sections were pre-treated for 3 min in absolute ethanol, followed by 1 min in 70% ethanol and 1 min in distilled water. They were then oxidized in a solution of 0.06% KMnO4 for 15 min. After three rinses of 1 min each in distilled water, the sections were incubated for 30 min in a solution of 0.001% Fluoro-Jade B (Chemicon) containing 0.01% of DAPI (Sigma) in 0.1% acetic acid. The slides were rinsed in deionized water for 3 min each, dried overnight, cleared in xylene, cover-slipped with Eukitt (Merck, Darmstadt, Germany) and examined using an Olympus (Hamburg, Germany) BX61 epifluorescence microscope. The statistical analysis of the obtained data was performed using Bonferroni post hoc test (Multiple comparison test) using Prism 5.0c (Mac OsX, Grahpad). Data are presented as mean ± standard error of the mean (S.E.M.). A value of ***P < 0.01 was considered statistically significant.
Histology and immunofluorescence
For histology, mice were perfused with phosphate buffered 4% paraformaldehyde, pH 7.3 24 hours after the first KA injection, post-fixed overnight in the same fixative and cryoprotected in 30% sucrose as above. A freezing microtome (Leica, Wetzlar, Germany) was used to obtain 30 μm-thick coronal sections, which were rinsed in 0.1 M PBS before 1 hour’s incubation at room temperature in 0.1 M PBS containing 0.2% gelatin, 10% normal goat serum, 0.2% glycine and 0.2% Triton X-100. Sections were then incubated overnight at 4 °C with indicated primary antibodies. After washing in 0.1 M PBS containing 0.2% Triton X-100, sections were incubated with goat anti-rabbit Alexa Fluor 568-tagged secondary antibody (1:200 diluted; Molecular Probes, Eugene, OR, USA), washed in 0.1 M, PBS and mounted in Fluoromount (Vector Labs, Burlingame, CA, USA). Immunohistochemical controls, which included omission or substitution of primary anti-GFAP antibody by either normal rabbit serum prevented immunostaining. For quantification of GFAP-positive astrocytes in the stratum radiatum of the dorsal hippocampal region, immunoreacted sections (5 sections of each mouse, n = 5 mice per genotype) were photodocumented with an Olympus BX61 fluorescence microscope equipped with a cooled DP12L camera. Photomicrographs were obtained using a 40X objective with identical time exposure (100–150 ms) between preparations from each wild-type and respective knockout mouse. No modifications were applied to the obtained pictures. Numbers of GFAP-expressing cells were determined by counting positive cells in five frames (250 × 200 μm) corresponding to the hippocampal CA1-3 regions of five mice of each genotype. Data were expressed as mean ± standard error of the mean (S.E.M). The statistical analysis of the obtained data was performed using Mann–Whitney U non-parametric test using Prism 5.0c (Mac OsX, Grahpad). A value of P < 0.01 was considered statistically significant.
RT-qPCR
Total RNA from hippocampal samples obtained from treated (6 h after KA-administration) and non-treated mice was purified with the mirVana isolation kit (Ambion, Austin, TX, USA) and used to make the single-stranded cDNAs required as templates for the RT-qPCR amplification. Sets of primers used in this study were: for TNFα 5′- AGCAAACCACCAAGTGGAGGA- 3′ and 5′-GCTGGCACCACTAGTTGGTTGT- 3′; and for ILβ 5′- TTGTGGCTGTGGAGAAGCTGT- 3′ and 5′- AACGTCACACACCAGCAGGTT- 3′. The reaction was performed with the Roche LightCycler 480 detector, using 2x SYBR Green Master Mix (Roche) as reagent, as indicated by the manufacturer. Amplification protocol consisted of a denaturation-activation cycle (95 °C for 10 min) followed by 40 cycles of denaturation-annealing-extension (95 °C, 15 sec; 60 °C, 40 sections; 72 °C, 5 sec; 98 °C, continuous). LightCycler 480 software was used for mRNA quantification. The data were analysed using the ΔΔCt method, which provides the target gene expression values as fold changes in the problem sample compared with a calibrator sample. Both problem and calibrator samples were normalized by the relative expression of a housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]). These analyses developed 3 different samples. The statistical analysis of the obtained data was performed using Mann–Whitney U non-parametric test using Prism 5.0c (Mac OsX, Grahpad). Data are presented as mean ± standard error of the mean (S.E.M.). A value of ***P < 0.01 was considered statistically significant.
Cell culture and treatments
The murine neuroblastoma cell line Neuro2a (N2a) expressing low levels of PrPC was grown at 37 °C, 5.5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4.5 g/L glucose, 10% fetal bovine serum (FBS) and antibiotics (Invitrogen-Life Technologies, Barcelona, Spain). For vector transfection, cells were plated at 1 × 105 cells/well in a 24-well plate and transiently transfected the next day using Lipofectamine 2000 reagent in Optimem medium, as indicated by the manufacturer (Invitrogen-Life Technologies). Four hours after transfection, cells were washed and the medium was replaced with DMEM containing 10% FBS. PrPC expression after transfection was checked with western blot analysis. In a first set of experiments, N2a cell cultures were transiently transfected with pcDNA and pcDNA-PrPC and maintained in vitro at 37 °C, 5.5% CO2. Cells were deprived for 24 h and treated with Glimepiride 20 μM and 0.2 u/ml PLC (final concentration). We used both treatments since some reports indicate that Glimepiride may act on ATP-dependent K+ channels that may also affect cell survival91. One hour later, 5 mM KA dissolved in 0.1 M PBS was added to the media. After treatment, cultures were rinsed twice in KA-free culture medium and cell viability was determined with WST-1 viability assay (see below). In parallel, non-permeabilized cells were processed to PrPC detection by immunofluorescence using the 6H4 antibody and a goat anti-mouse Alexa Fluor 488–tagged secondary antibody (1:200 diluted; Molecular Probes). For fluorescence quantification of cell-bound Alexa Fluor 488, immunoreacted cultures (n = 10 per experimental group) were photodocumented with an Olympus BX61 + DP12L camera. Photomicrographs were obtained using a 20X objective with identical time exposure (250–300 ms) for preparations from each group. No modifications were applied to the obtained pictures. Fluorescence intensity was determined using ImageJ by measuring the corrected total cell fluorescence (CTCF) as: CTCF = integrated density – (area of selected N2a mesured cells x mean fluorescence of background). Data were expressed as mean ± standard error of the mean (S.E.M). The statistical analysis of the obtained data in these experiments was performed using Bonferroni post hoc test (Multiple comparison test) using Prism 5.0c (Mac OsX, Grahpad). A value of P < 0.05 was considered statistically significant. These experiments were repeated four times. In a second set of experiments, N2a cells were transfected with pcDNA3.1, pcDNA3.1-PrPC, pcDNA3.1-PrP∆CD and pcDNA3.1-PrP∆F35. KA (5 mM) treatment was carried out on serum-deprived cells 24 h after transfection. Cell viability was determined using a commercially available WST-1-based assay. Cell cultures were incubated with WST-1 reagent for 2 hours. Then absorbance at 450 nm was measured in a multiwell plate reader (Merck ELISA System MIOS). Data were normalized with A450 in untreated controls. pcDNA3.1-PrPC, pcDNA3.1-PrP∆CD and pcDNA3.1-PrP∆F35 were kind gifts from Prof. D. Harris (Boston University) and Prof. A. Aguzzi (University Hospital of Zurich). These experiments were repeated five times. The statistical analysis of the obtained data in these experiments was performed using Bonferroni post hoc test (Multiple comparison test) using Prism 5.0c (Mac OsX, Grahpad). Data are presented as mean ± standard error of the mean (S.E.M.). Values of **P < 0.05 and ***P < 0.01 were considered statistically significant.
Additional Information
How to cite this article: Carulla, P. et al. Involvement of PrPC in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 5, 11971; doi: 10.1038/srep11971 (2015).
References
Steele, A. D., Emsley, J. G., Ozdinler, P. H., Lindquist, S. & Macklis, J. D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci USA 103, 3416–3421 (2006).
Llorens, F. et al. PrP(C) regulates epidermal growth factor receptor function and cell shape dynamics in Neuro2a cells. J Neurochem 127, 124–138 (2013).
Brown, D. R. et al. The cellular prion protein binds copper in vivo. Nature 390, 684–687 (1997).
Vassallo, N. & Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J Neurochem 86, 538–544 (2003).
Brown, D. R., Nicholas, R. S. & Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 67, 211–224 (2002).
Mouillet-Richard, S. et al. Signal transduction through prion protein. Science 289, 1925–1928 (2000).
Hugel, B. et al. Modulation of signal transduction through the cellular prion protein is linked to its incorporation in lipid rafts. Cell Mol Life Sci 61, 2998–3007 (2004).
Bueler, H. et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582 (1992).
Manson, J. C. et al. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8, 121–127 (1994).
Steele, A. D., Lindquist, S. & Aguzzi, A. The prion protein knockout mouse: a phenotype under challenge. Prion 1, 83–93 (2007).
Gadotti, V. M., Bonfield, S. P. & Zamponi, G. W. Depressive-like behaviour of mice lacking cellular prion protein. Behav Brain Res 227, 319–323 (2012).
Criado, J. R. et al. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis 19, 255–265 (2005).
Schmitz, M. et al. Loss of Prion Protein Leads to Age-Dependent Behavioral Abnormalities and Changes in Cytoskeletal Protein Expression. Mol Neurobiol (2014).
Le Pichon, C. E. et al. Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nat Neurosci 12, 60–69 (2009).
Bremer, J. et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13, 310–318 (2010).
Tobler, I. et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 380, 639–642 (1996).
Walz, R. et al. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 40, 1679–1682 (1999).
Rangel, A. et al. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis and death: Role of AMPA/kainate receptors. J Neurosci Res 85, 2741–2755 (2007).
Khosravani, H. et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Gen Physiol 131, i5 (2008).
Carulla, P. et al. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol Biol Cell 22, 3041–3054 (2011).
Llorens, F. & Del Rio, J. A. Unraveling the neuroprotective mechanisms of PrP (C) in excitotoxicity. Prion 6, 245–251 (2012).
Striebel, J. F., Race, B. & Chesebro, B. Prion protein and susceptibility to kainate-induced seizures: genetic pitfalls in the use of PrP knockout mice. Prion 7, 280–285 (2013).
Striebel, J. F., Race, B., Pathmajeyan, M., Rangel, A. & Chesebro, B. Lack of influence of prion protein gene expression on kainate-induced seizures in mice: studies using congenic, coisogenic and transgenic strains. Neuroscience 238, 11–18 (2013).
Ratte, S. et al. Threshold for epileptiform activity is elevated in prion knockout mice. Neuroscience 179, 56–61 (2011).
Schauwecker, P. E. & Steward, O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 94, 4103–4108 (1997).
McKhann, G. M., 2nd, Wenzel, H. J., Robbins, C. A., Sosunov, A. A. & Schwartzkroin, P. A. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality and hippocampal pathology. Neuroscience 122, 551–561 (2003).
Schauwecker, P. E. Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp Neurol 178, 219–235 (2002).
Schauwecker, P. E. Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res 135, 139–148 (2002).
McCord, M. C., Lorenzana, A., Bloom, C. S., Chancer, Z. O. & Schauwecker, P. E. Effect of age on kainate-induced seizure severity and cell death. Neuroscience 154, 1143–1153 (2008).
Nuvolone, M. et al. SIRPalpha polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J Exp Med 210, 2539–2552 (2013).
Bribian, A. et al. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PloS One 7, e33872 (2012).
Gerlai, R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci 19, 177–181 (1996).
Smithies, O. & Maeda, N. Gene targeting approaches to complex genetic diseases: atherosclerosis and essential hypertension. Proc Natl Acad Sci U S A 92, 5266–5272 (1995).
Herms, J. W., Kretzchmar, H. A., Titz, S. & Keller, B. U. Patch-clamp analysis of synaptic transmission to cerebellar purkinje cells of prion protein knockout mice. Eur J Neurosci 7, 2508–2512 (1995).
Lledo, P. M., Tremblay, P., DeArmond, S. J., Prusiner, S. B. & Nicoll, R. A. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci USA 93, 2403–2407 (1996).
Mercer, R. C. et al. The prion protein modulates A-type K+ currents mediated by Kv4.2 complexes through dipeptidyl aminopeptidase-like protein 6. J Biol Chem 288, 37241–37255 (2013).
Collinge, J. et al. Prion protein is necessary for normal synaptic function. Nature 370, 295–297 (1994).
Manson, J. C. et al. PrP gene dosage and long term potentiation. Neurodegeneration 4, 113–114 (1995).
Whittington, M. A. et al. Rescue of neurophysiological phenotype seen in PrP null mice by transgene encoding human prion protein. Nature Genet 9, 197–201 (1995).
Rangel, A. et al. Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PloS One 4, e7592 (2009).
Fournier, J. G. Cellular prion protein electron microscopy: attempts/limits and clues to a synaptic trait. Implications in neurodegeneration process. Cell Tissue Res 332, 1–11 (2008).
Brown, D. R. Prion and prejudice: normal protein and the synapse. Trends Neurosci 24, 85–90 (2001).
Herms, J. et al. Evidence of presynaptic location and function of the prion protein. J Neurosci 19, 8866–8875 (1999).
Keshet, G. I., Bar-Peled, O., Yaffe, D., Nudel, U. & Gabizon, R. The cellular prion protein colocalizes with the dystroglycan complex in the brain. J Neurochem 75, 1889–1897 (2000).
Spielhaupter, C. & Schatzl, H. M. PrPC directly interacts with proteins involved in signaling pathways. J Biol Chem 276, 44604–44612 (2001).
Watt, N. T. et al. Prion protein facilitates uptake of zinc into neuronal cells. Nature Commun 3, 1134 (2012).
Beraldo, F. H. et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J 25, 265–279 (2011).
Gasperini, L., Meneghetti, E., Pastore, B., Benetti, F. & Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 22, 772–84 (2015).
Royle, S. J., Collins, F. C., Rupniak, H. T., Barnes, J. C. & Anderson, R. Behavioural analysis and susceptibility to CNS injury of four inbred strains of mice. Brain Res 816, 337–349 (1999).
Schauwecker, P. E. Genetic basis of kainate-induced excitotoxicity in mice: phenotypic modulation of seizure-induced cell death. Epilepsy Res 55, 201–210 (2003).
Chesebro, B. et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1435–1439 (2005).
Bate, C., Tayebi, M., Diomede, L., Salmona, M. & Williams, A. Glimepiride reduces the expression of PrPc, prevents PrPSc formation and protects against prion mediated neurotoxicity in cell lines. PloS One 4, e8221 (2009).
Shmerling, D. et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93, 203–214 (1998)
Flechsig, E. et al. Expression of truncated PrP targeted to Purkinje cells of PrP knockout mice causes Purkinje cell death and ataxia. EMBO J 22, 3095–3101 (2003).
Nicolas, O. et al. Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. FASEB J 21, 3107–3117 (2007).
Maglio, L. E., Martins, V. R., Izquierdo, I. & Ramirez, O. A. Role of cellular prion protein on LTP expression in aged mice. Brain Res 1097, 11–18 (2006).
Curtis, J., Errington, M., Bliss, T., Voss, K. & MacLeod, N. Age-dependent loss of PTP and LTP in the hippocampus of PrP-null mice. Neurobiol Dis 13, 55–62 (2003).
Crusio, W. E. Flanking gene and genetic background problems in genetically manipulated mice. Biol Psychiatry 56, 381–385 (2004).
Ferraro, T. N. et al. Mouse strain variation in maximal electroshock seizure threshold. Brain Res 936, 82–86 (2002).
Jackson, C., Welch, H. C. & Bellamy, T. C. Control of cerebellar long-term potentiation by P-Rex-family guanine-nucleotide exchange factors and phosphoinositide 3-kinase. PLoS One 5, e11962 (2010).
Wang, L. et al. SHPS-1 deficiency induces robust neuroprotection against experimental stroke by attenuating oxidative stress. J Neurochem 122, 834–843 (2012).
Lu, Y. Y. et al. TRAF1 is a critical regulator of cerebral ischaemia-reperfusion injury and neuronal death. Nat Commun 4, 2852 (2013).
Hennekinne, L., Colasse, S., Triller, A. & Renner, M. Differential control of thrombospondin over synaptic glycine and AMPA receptors in spinal cord neurons. J Neurosci 33, 11432–11439 (2013).
Okada-Tsuchioka, M. et al. Electroconvulsive seizure induces thrombospondin-1 in the adult rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry 48, 236–244 (2014).
Petri, M. K. et al. PTPIP51, a positive modulator of the MAPK/Erk pathway, is upregulated in glioblastoma and interacts with 14-3-3beta and PTP1B in situ. Histol Histopathol 26, 1531–1543 (2011).
Ji, R. et al. TAM receptors affect adult brain neurogenesis by negative regulation of microglial cell activation. J Immunol 191, 6165–6177 (2013).
Li, Q., Lu, Q., Lu, H. & Tian, S. Systemic autoimmunity in TAM triple knockout mice causes inflammatory brain damage and cell death. PLoS One 8, e64812 (2013).
Aguilar-Alonso, P. et al. The increase in zinc levels and upregulation of zinc transporters are mediated by nitric oxide in the cerebral cortex after transient ischemia in the rat. Brain Res 1200, 89–98 (2008).
Neher, J. J. et al. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 110, E4098–4107 (2013).
Nelson, P. A. et al. MHC class I immune proteins are critical for hippocampus-dependent memory and gate NMDAR-dependent hippocampal long-term depression. Learn Mem 20, 505–517 (2013).
Fleisch, V. C. et al. Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol Dis 55, 11–25 (2013).
Baumann, F. et al. Functionally relevant domains of the prion protein identified in vivo. PloS One 4, e6707 (2009).
Sonati, T. et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106 (2013).
Herrmann, U. S. et al. Prion Infections and Anti-PrP Antibodies Trigger Converging Neurotoxic Pathways. PLoS Pathog 11, e1004662 (2015).
Zeng, F., Watt, N. T., Walmsley, A. R. & Hooper, N. M. Tethering the N-terminus of the prion protein compromises the cellular response to oxidative stress. J Neurochem 84, 480–490 (2003).
Dupiereux, I. et al. Protective effect of prion protein via the N-terminal region in mediating a protective effect on paraquat-induced oxidative injury in neuronal cells. J Neurosci Res 86, 653–659 (2008).
Watts, J. C. & Westaway, D. The prion protein family: diversity, rivalry and dysfunction. Biochem Biophys Acta 1772, 654–672 (2007).
Linden, R. et al. Physiology of the prion protein. Physiol Rev 88, 673–728 (2008).
Pauly, P. C. & Harris, D. A. Copper stimulates endocytosis of the prion protein. J Biol Chem 273, 33107–33110 (1998).
Mitteregger, G. et al. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol 17, 174–183 (2007).
Sakudo, A. et al. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem Biophys Res Commun 308, 660–667 (2003).
Taylor, D. R., Watt, N. T., Perera, W. S. & Hooper, N. M. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci 118, 5141–5153 (2005).
Martens, U., Capito, B. & Wree, A. Septotemporal distribution of [3H]MK-801, [3H]AMPA and [3H]Kainate binding sites in the rat hippocampus. Anat Embryol (Berl) 198, 195–204 (1998).
Ong, W. Y., Leong, S. K., Garey, L. J., Reynolds, R. & Liang, A. W. An immunocytochemical study of glutamate receptors and glutamine synthetase in the hippocampus of rats injected with kainate. Exp Brain Res 109, 251–267 (1996).
Hampson, D. R. et al. Localization of AMPA receptors in the hippocampus and cerebellum of the rat using an anti-receptor monoclonal antibody. Neuroscience 50, 11–22 (1992).
Baumann, F. et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 26, 538–547 (2007).
Klohn, P. C. et al. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science 335, 52 (2012).
Solforosi, L. et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303, 1514–1516 (2004).
Flechsig, E. et al. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 27, 399–408 (2000).
Slotnick, B. M. & Leonard, C. M. A stereotaxic atlas of the albino mouse forebrain. (U.S. Dept. of Health, Education and Welfare, Public Health Service, Alcohol, Drug Abuse for sale by the Supt. of Docs., U.S. Govt. Print. Off., 1975).
Mathew, A. J., Raj, N. N., Sugappriya, M. & Priyadarshini, S. M. Modeling of ATP-sensitive inward rectifier potassium channel 11 and inhibition mechanism of the natural ligand, ellagic acid, using molecular docking. Adv Exp Med Biol 680, 489–495 (2010).
Acknowledgements
The authors thank Tom Yohannan for editorial advice and M. Segura for technical assistance. We thank Jesús R. Requena (CIMUS, Santiago de Compostela, Spain) for helpful comments on the manuscript. We also thank members of the Del Río, Torres, Ferrer and Legname groups for stimulating discussions and ideas. This research was supported by the Spanish Ministry of Science and Innovation (BFU2012-32617), FP7-PRIORITY, the Generalitat de Catalunya (SGR2014-1218), CIBERNED (PI2014/02-4 and PRY-14-114), La Caixa Obra Social Foundation, La Marató de TV3 and the Basque Foundation of Health and Innovation Research (BIO12/AL/004) to JADR. R.G. was supported by Insituto carlos Tercero (FIS, PI11-00075). I.F. was supported by FIS (PI1100968) and CIBERNED. JM.T. was supported by the Spanish Ministry of Science and Innovation (RTA2012-00004 and AGL2012-37988-C04 projects) and EU FP7 project ‘Priority CT2009-222887’. G.L.’s funding was provided by the EU FP7 project ‘Priority CT2009-222887’. P.C. was supported by AGAUR and A.M-A. was supported by a fellowship from the Fundación Tatiana Pérez de Guzmán el Bueno. P. A-C. was supported by a fellowship from the Spanish Ministry of Science and Innovation (BES-2010-349 040922).
Author information
Authors and Affiliations
Contributions
P.C., F.Ll., A.M-A., P.A.-C., R.G. and J.C.E. performed most of the experiments. I.F., J.M.T., G.L. and J.A.D.R. designed the project and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Carulla, P., Llorens, F., Matamoros-Angles, A. et al. Involvement of PrPC in kainate-induced excitotoxicity in several mouse strains. Sci Rep 5, 11971 (2015). https://doi.org/10.1038/srep11971
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11971
This article is cited by
-
Analysis of co-isogenic prion protein deficient mice reveals behavioral deficits, learning impairment, and enhanced hippocampal excitability
BMC Biology (2022)
-
The Prion Protein Regulates Synaptic Transmission by Controlling the Expression of Proteins Key to Synaptic Vesicle Recycling and Exocytosis
Molecular Neurobiology (2019)
-
The function of the cellular prion protein in health and disease
Acta Neuropathologica (2018)
-
Involvement of Cellular Prion Protein in α-Synuclein Transport in Neurons
Molecular Neurobiology (2018)
-
The biological function of the cellular prion protein: an update
BMC Biology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
