
Abstract
RalA and RalB proteins are key mediators of oncogenic Ras signaling in human oncogenesis. Herein we investigated the mechanistic contribution of Ral proteins to invasion of lung cancer A549 cells after induction of epithelial-mesenchymal transition (EMT) with TGFβ. We show that TGFβ-induced EMT promotes dissemination of A549 cells in a 2/3D assay, independently of proteolysis, by activating the Rho/ROCK pathway which generates actomyosin-dependent contractility forces that actively remodel the extracellular matrix, as assessed by Traction Force microscopy. RalB, but not RalA, is required for matrix deformation and cell dissemination acting via the RhoGEF GEF-H1, which associates with the Exocyst complex, a major Ral effector. Indeed, uncoupling of the Exocyst subunit Sec5 from GEF-H1 impairs RhoA activation, generation of traction forces and cell dissemination. These results provide a novel molecular mechanism underlying the control of cell invasion by RalB via a cross-talk with the Rho pathway.
Similar content being viewed by others


Introduction
In the last 15 years Ral signaling pathway came to the front of the stage as a major player in human oncogenesis1,2,3. The small GTPase proteins RalA and RalB act downstream of the Ras oncoproteins; indeed, the direct Ral activators (RalGEFs, Guanine Exchange Factors) are direct targets of active Ras. Ral proteins have been involved in a variety of cellular processes (such as motility/invasion, apoptosis, cytokinesis and autophagy) and in various steps of tumor development (including tumor formation, survival, growth and metastasis). Some of these functions are shared by the two Rals, others are distinct. In particular, in cancer, RalA was found to be necessary for the anchorage-independent growth of tumor cells, while RalB was shown to be critical for survival4, for motility5,6 and for metastasis7,8.
Concerning the role of Ral in cell migration, our laboratory showed that RalB promotes assembly of the Exocyst complex, a major Ral effector and its localization to the leading edge of motile cells6. The Exocyst complex, composed of eight subunits (Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70 and Exo84), mediates the targeting and tethering of post-Golgi secretory vesicles to specific membrane sites during polarized exocytosis9. Exocyst is molecularly connected to a Rac1 negative regulator (the RacGAP SH3BP1, GTPase Activating Protein)10 and to a Rac1 effector (the Wave Regulatory complex) (Biondini et al, manuscript in preparation). These works indicated that the RalB/Exocyst pathway contributes to spatially organize the signaling of the Rac1 GTPase, which is a master regulator of actin polymerization in lamellipodia.
For migration through an extra-cellular matrix (ECM), cells apply several invasion strategies (mesenchymal and amoeboid, single-cell and collective) which imply degradation and/or deformation of the ECM11. The choice among the various invasion strategies relies not only on the cell autonomous characteristics (such as genetic features), but also on the physical, cellular and biochemical properties of the tumor environment12,13. Among the many pro-invasive soluble factors present in the tumor environment, the transforming growth factor β (TGFβ) plays multi-facet roles which still await for a better comprehension in order to develop effective anti-metastasis therapies14,15. More specifically, TGFβ can promote tumorigenesis by activating the epithelial-mesenchymal transition (EMT) program16. EMTs physiologically take place during embryo morphogenesis allowing the conversion of stationary epithelial cells into mesenchymal cells able to migrate and invade. In the past decade, an increasing number of ex vivo and in vivo studies has provided evidences for the recapitulation of the EMT program during metastasis17.
In this work we aimed at understanding the interplay between two major contributors to cancer cell dissemination: the Ral pathway and the TGFβ-induced EMT. We used as cellular model the lung adenocarcinoma A549 cells that undergo a full EMT upon TGFβ treatment18. We found that for invasion in 2-3D Matrigel, in presence of TGFβ, A549 cells opt for a contractility-driven strategy without proteolytic degradation of the ECM, but instead with generation of traction forces on the ECM. In these conditions, we showed that the RalB/Exocyst signaling axis plays an essential role in controlling contractility by interacting with the RhoGEF GEF-H1, which stimulates the Rho-ROCK-MLC2 pathway.
Results
TGFβ-induced EMT promotes protease-independent dissemination of A549 cells in 2-3D
The lung adenocarcinoma A549 cells are a well-established model for ex vivo EMT studies. They undergo progressive EMT upon TGFβ treatment18,19. We confirmed that TGFβ treatment for 4 days induced EMT both by morphological changes (Supplementary Figure S1A) and by a switch from epithelial to mesenchymal markers (Supplementary Figure S1B). To allow the full acquisition of EMT-related invasive behaviors, we routinely treated the cells with TGFβ for at least 7 days, which allowed a complete rearrangement of actin cytoskeleton and adhesion sites to the substrate (Supplementary Figure S1C).
To study invasion we took advantage of the Circular Invasion Assay (CIA)20 because it allows detailed time-lapse microscopic analysis during the invasion process (Fig. 1A). In this assay the cells are in contact with the matrix (Matrigel) both at the dorsal and frontal sides but they ventrally adhere to the bottom-glass dish; therefore it is a 2-3D situation which mimics a topography that is often observed during cancer invasion in vivo21. We compared the invasive phenotype of untreated versus TGFβ-treated cells. Untreated cells invaded through the matrix in a collective-like manner, keeping cell-cell coordination and with a rather compact front, but they were never able to invade deeply during the 2-day observation time (Fig. 1B and Supplementary Movie S1). On the contrary, TGFβ-treated cells displayed more chaotic dynamics inside the monolayer; some cells were detaching from the group and disseminating through the matrix till distant sites (Fig. 1B and Supplementary Movie S1). By single-cell tracking analysis we measured a “dissemination speed”, defined as the velocity of the cell movements in this 2-3D setting. The dissemination speed was almost 2-fold higher in TGFβ-treated cells (12.74 μm/hr ± 0.51 SEM) as compared to untreated cells (7.09 μm/hr ± 0.56 SEM) (Fig. 1C). We concluded that, in 2-3D Matrigel environment, TGFβ-induced EMT increases A549 cell invasion and promotes a shift from collective to single-cell dissemination.

Dissemination of TGFβ-treated A549 cells in 2/3D is proteolysis independent.
(A) Depiction of the 2/3D Circular Invasion Assay (CIA). Note that we kept the original name of the assay, but that the actual shape of the stopper we used is not circular but square. (B) EMT promotes cell dissemination. A549 cells were treated with 2 ng/mL TGFβ for 7 days, submitted to CIA and compared to untreated cells. Selected time points from a representative experiment are shown. See Supplementary Movie S1 for entire video sequence. Scale bar, 100 μm. (C) Quantification of cell dissemination. Individual cells were tracked using the ImageJ software. Number of cells n = 40 for untreated and n = 163 for TGFβ-treated conditions from at least four experiments per condition. (D) TGFβ greatly stimulates invadopodia formation. Cells were stimulated with TGFβ for 7 days and invadopodia were visualized as dots positive for matrix degradation (black), for cortactin staining (green) and for F-actin staining (red). Scale bar, 20 μm.(E) Quantification of cells positive for at least one invadopodia. Counting was performed on three independent experiments (n≥100 cells/condition per experiment). (F) TGFβ induces strong secretion of MMP2 and MMP9 metalloproteinases. Conditioned media from untreated and TGFβ-treated cells were collected and processed for gelatin zymogram assay. A representative zymogram image is shown. (G) MMP-dependent proteolysis is dispensable for TGFβ-induced dissemination in 2/3D. TGFβ-treated cells were incubated with 25 μM GM6001 MMPs inhibitor (2 hrs before CIA and during CIA) or depleted of MMP2 and MMP9 and submitted to CIA. Number of cells n>30 per condition from three experiments with GM6001 and one experiment with siRNAs. Error bars represent SEM. p values come from two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significant.
The TGFβ treatment greatly stimulated invadopodia formation (Fig. 1D,E) and metalloproteinase secretion (Fig. 1F), as expected from previous reports22,23. However, very surprisingly, the dissemination speed in presence of TGFβ was not affected by the addition of a pan-inhibitor of metalloproteinases (GM6001) nor by the depletion of two major secreted metalloproteinases (MMP2 and MMP9) (Fig. 1G and S2A), indicating that TGFβ-induced invasion in 2-3D may occur via a protease-independent mechanism.
TGFβ-treated A549 cells disseminate by a RhoA-dependent matrix deformation mechanism
A careful inspection of the videos of disseminating TGFβ-treated A549 cells in CIA revealed that they were remodeling the surrounding Matrigel matrix and leaving behind dark shadows of matrix deformation in their paths (Supplementary Movie S2). Similarly, when seeded on a thick layer of collagen gel, TGFβ-treated A549 cells clearly pulled the underlying gel in a very dynamic way, contrary to untreated cells (Supplementary Movie S3). Therefore we hypothesized that their dissemination strategy could depend on forces generated by actomyosin contractility.
We took advantage of a recent biophysical technology, the Traction Force Microscopy (TFM), to provide spatially-resolved measurements of interfacial forces through the quantification of the deformation of an elastic substrate by the cells24. By this approach we measured the “strain energy”, which is the total energy transferred from a cell to the elastic distortion of the substrate. The strain energy of TGFβ-treated A549 cells was almost 2-fold higher than that of untreated A549 cells (0.18 pJ ± 0.02 SEM versus 0.1 pJ ± 0.01 SEM, respectively) (Fig. 2A,B). This finding indicates that TGFβ-treated A549 cells were applying much stronger traction forces on the substrate as compared to untreated A549 cells.

TGFβ1-induced EMT enhances actomyosin contractility and matrix remodelling.
(A) EMT promotes traction forces on the substratum. Representative phase contrast images and corresponding traction force maps of untreated and TGFβ-treated A549 cells as revealed by Traction Force Microscopy (TFM). Color scale bar denotes traction stress (Pa). Scale bar, 50 μm. (B) Quantification of the strain energy. TGFβ-treated cells generate significantly higher strain energies compared to untreated cells. Number of untreated cells n = 51, TGFβ-treated cells n = 35, from two experiments. (C) EMT stimulates Rho activity. Visualization of RhoA activity in representative untreated and TGFβ-treated cells. Cells were transfected with a plasmid expressing Raichu-RhoA (KRasCter) biosensor and observed by FRET microscopy the day after. Note the higher level of RhoA at cell periphery. Scale bars, 20 μm. (D) Quantification of whole-cell RhoA activity. YFP and CFP images were acquired for each cell and mean intensities were measured for the entire cell surface. The YFP/CFP ratio is a measure of whole-cell FRET, i.e. of whole-cell RhoA activity. Number of untreated cells n = 54, TGFβ-treated cells n = 41, from two experiments. (E) EMT induces MLC2 phosphorylation. The levels of total and phosphorylated MLC2 were analyzed by western-blot. The quantification of band intensities (with respect to untreated condition) and the calculated P-MLC/MLC ratio are indicated. The vertical lanes indicate positions were gel images were cut in order to juxtapose non-adjacent lanes coming from the same gel. (F) EMT stimulates ROCK-dependent actomyosin contractility. A549 cells were grown with or without TGF-β1 for 7 days and embedded in collagen I gels for 4 days. Gel contraction was monitored. Where indicated, cells were pre-treated for 2 hrs with 10 μM Y27632 ROCK inhibitor and submitted to collagen contraction assay in presence of the inhibitor. Number of untreated gels n = 2, TGFβ-treated gels n = 2, TGFβ/Y27632-treated gels n = 1, from two experiments. Error bars represent SEM. p values come from two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.Scale bar is 100 μm.
The RhoA pathway regulates actomyosin contractility via the ROCK kinase that phosphorylates the myosin light chain 2 (MLC2)25. In TGFβ-treated cells the RhoA activity was higher than in untreated cells as measured by a FRET-based biosensor (Fig. 2C,D). Consistently, the phosphorylation of MLC2 was strongly increased; noteworthy, also the level of total MLC2 was slightly increased in TGFβ-treated cells (Fig. 2E). Finally, by collagen gel contraction assays, we found that the matrix remodeling capacity of TGFβ-treated cells was much higher than that of untreated cells (Fig. 2F). As expected, the addition of the ROCK inhibitor Y27632 impaired MLC2 phosphorylation (Fig. 2E) and gel contractility (Fig. 2F).
All these data together support the conclusion that TGFβ-induced EMT increases force generation capability of A549 cells through the RhoA-ROCK-MLC2 pathway, therefore promoting dissemination in 2-3D via a contractility-dependent matrix deformation mechanism.
RalB, but not RalA, is required for the dissemination of TGFβ-treated A549 cells
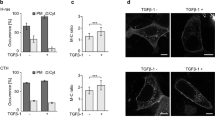
We next investigated the role of Ral proteins in TGFβ-induced dissemination of A549 cells. TGFβ-treated cells were depleted of RalA or RalB by respectively 2 and 3 independent siRNAs and analyzed by Circular Invasion Assay (CIA). The silencing of RalB significantly impaired dissemination speed, while the silencing of RalA had no effect (Fig. 3A). The depletion levels were ≥80% (Fig. 3B). We verified that Ral depletion does not impair proliferation of TGFβ-treated A549 cells (Supplementary Figure S3A). Anyway, the strategy of tracking cells one-by-one allows us to exclude possible indirect effects of siRNA treatments on cell division and/or survival in the determination of dissemination speeds.

RalB regulates dissemination of TGFβ-treated cells.
(A) RalB silencing impairs TGFβ-induced dissemination in CIA. TGFβ-treated cells were depleted of RalA or RalB, submitted to CIA and the speed of dissemination was evaluated by manual tracking. Number of cells from at least three experiments: siControl n = 76, siRalA-I n = 66, siRalA-II n = 44, siRalB-107 n = 30, siRalB-1749 n = 13, siRalB-333 n = 28. (B) Validation of Ral proteins depletion. Efficiency of RalA and RalB depletion was verified by western blotting.(C) Effect of Ral depletion on TGFβ-induced contractility. Representative images showing gel contraction by TGFβ-treated cells depleted of RalA or RalB. (D) Quantification of gel contraction upon Ral depletion. Mean percentage values from four to five experiments were plotted on graphic. Error bars represent SEM. p values come from two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
In addition, silencing of RalB, but not RalA, significantly impaired gel contraction capacity of TGFβ-treated cells (Fig. 3C,D). We also found that depletion of RalA or RalB does not impair secretion of MMP2 and MMP9 metalloproteinases, arguing again against a protease-dependent invasion mechanism of TGFβ-treated cells in CIA (Supplementary Figure S2B).
In conclusion, RalB but not RalA is required to regulate contractility-driven dissemination of A549 cells upon TGFβ stimulation.
The Ral pathway mobilizes the RhoGEF GEF-H1 to drive TGFβ-induced dissemination
How RalB controls TGFβ-induced dissemination in 2-3D? Pathak and colleagues had previously shown a connection between the Ral pathway and the Rho pathway26. They reported that GEF-H1, a Guanine nucleotide Exchange Factor (GEF) for Rho proteins, directly binds to the Sec5 subunit of the Exocyst complex in a Ral-dependent manner. In addition, the GEF-H1/RhoA signaling activity has been linked to several processes such as cytokinesis27, mechano-signaling28,29, cancer motility, invasion and metastasis30,31,32,33,34,35,36. We reasoned that RalB could regulate TGFβ-induced dissemination by controlling Rho-driven actomyosin contractility via the Exocyst/GEF-H1 interaction.
First, we assessed the consequences of GEF-H1 depletion on dissemination and contractility of TGFβ-treated cells in our settings. Silencing of GEF-H1 by any of 2 siRNAs decreased dissemination speed (Fig. 4A) and reduced gel contraction capacity (Fig. 4C,D), despite a weak ~50% depletion efficiency (Fig. 4B). Noteworthy, GEF-H1 depletion had no significant effects on proliferation of TGFβ-treated A549 cells (Supplementary Figure S3B). These results indicate the necessity of the RhoGEF GEF-H1 for TGFβ-induced dissemination in 2-3D.

GEF-H1 controls both dissemination and contractility of TGFβ-treated cells.
(A) GEF-H1 depletion impairs TGFβ-induced dissemination in CIA. TGFβ-treated cells were depleted of GEF-H1 and subsequently submitted to CIA. Impact of GEF-H1 silencing on dissemination speed was quantified by manual tracking. Number of cells from at least three experiments: siControl n = 81, siGEF-H1#2 n = 47, siGEF-H1#smart pool n = 32, (B) Validation of GEF-H1 protein depletion. Efficiency of GEF-H1 depletion was verified by western blotting. The vertical lanes indicate positions were gel images were cut in order to juxtapose non-adjacent lanes coming from the same gel. (C) Effect of GEF-H1 depletion on TGFβ-induced contractility. Representative images showing gel contraction by TGFβ-treated cells depleted of GEF-H1. (D) Quantification of gel contraction upon GEF-H1 depletion. Mean percentage values from two to six experiments were plotted on graphic.
Second, we perturbed the endogenous binding between the Sec5 Exocyst subunit and GEF-H1 by expressing a Cherry-fused GEF-H1aa119–236 competing peptide which corresponds to the minimal GEF-H1 domain interacting with Sec526. Previously, this strategy was successfully used to uncouple Sec5 from GEF-H1 and to show that the Exocyst/GEF-H1 interaction modulates vesicle trafficking26. By lentiviral infection, we stably expressed the Cherry-fused GEF-H1aa119–236 competing peptide and the Cherry control in TGFβ-treated A549 cells. Comparing to the control, the expression of GEF-H1aa119–236 led to decreased dissemination in CIA (Fig. 5A), to decreased RhoA activity (Fig. 5B) and to decreased capacity to generate traction forces (Fig. 5C,D), without impacting on cell proliferation (Supplementary Figure S3C). These results indicate that the binding of GEF-H1 to Exocyst is necessary to activate RhoA and to promote force-driven dissemination of TGFβ-treated cells.

Exocyst/GEF-H1 interaction is required for dissemination, RhoA activity and generation of traction forces of TGFβ-treated cells.
(A) Exocyst/GEF-H1 uncoupling impairs TGFβ-induced dissemination in CIA. TGFβ-treated cells stably expressing Cherry (control) or Cherry-GEF-H1aa119–236 (the peptide aa119–236 competes for the binding of endogenous GEF-H1 to Sec5) were submitted to CIA. Dissemination speeds were measured by manual tracking: Cherry n = 60, Cherry-GEF-H1aa119–236 n = 45, from two experiments. (B) Exocyst/GEF-H1 uncoupling perturbs RhoA activity. TGFβ-treated cells stably expressing Cherry or Cherry-GEF-H1aa119–236 were transfected with a FRET biosensor to visualize RhoA activity. Graphic shows mean whole-cell FRET values: Cherry n = 54, Cherry-GEF-H1aa119–236 n = 39, from two experiments. (C) Exocyst/GEF-H1 uncoupling impairs generation of traction forces. The ability of Cherry or Cherry-GEF-H1aa119–236 stably expressing cells to generate forces was investigated by Traction Force Microscopy (TFM). Phase contrast images and corresponding traction force maps are shown. Color scale bar denotes traction stress (Pa). Scale bar, 50 μm. (D) Quantification of the strain energy. Cherry-GEF-H1aa119–236 cells generate significantly less strain energies compared to Cherry. Number of Cherry cells n = 72, Cherry-GEF-H1aa119–236 cells n = 51, from two experiments. Error bars represent SEM. p values come from two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
These data strongly suggest that RalB, via its effector Exocyst, controls contractility-driven dissemination through the RhoGEF GEF-H1, which activates the RhoA-ROCK-MLC2 pathway and promotes traction forces.
Plasticity balance of RhoA and Rac1 pathways upon TGFβ-induced EMT
The above mentioned model predicts that inhibiting the ROCK kinase would inhibit dissemination in our settings. Surprisingly, despite a strong (expected) reduction of MLC2 phosphorylation (Fig. 2E), the number of disseminating cells appeared to increase unexpectedly in presence of the ROCK inhibitor Y27632 (Fig. 6A and Supplementary Movie S4). We counted the cells that in 2 days reached a distance >130 μm from the starting monolayer and we found an increase of almost 3-fold (Fig. 6B). We obtained similar results by direct inhibition of Rho proteins with C3 transferase (Supplementary Figure S4). These results are consistent with previous work that showed that RhoA depletion promotes invasive protrusions in MDA-MB-231 cells37. However, the single-cell dissemination speed of TGFβ-treated A549 cells was not affected by the addition of Y27632 (Fig. 6C, control versus Y27632). Interestingly, in presence of Y27632, cells displayed broad lamellipodia-like protrusions much more frequently than in control cells, suggesting that RhoA pathway inhibition may lead to Rac1 activation as a consequence of the well-known Rho/Rac biochemical balance37,38. We tested the activation status of Rac1 in TGFβ/Y27632-treated cells by employing a FRET-based Raichu-Rac1 biosensor. We observed an up-regulation of Rac1 activity, particularly at protrusion sites (Fig. 6D). The quantifications clearly showed that the addition of Y27632 stimulated the whole-cell Rac1 activity in TGFβ-treated cells (Fig. 6E).

Contractility inhibition leads to Rac activation and Rac-dependent dissemination.
(A) Inhibition of ROCK does not impair TGFβ-induced dissemination. TGFβ-treated cells were pre-treated for 2 hrs with 10 μM Y27632 ROCK inhibitor and then submitted to CIA in the presence of the inhibitor. Selected time points from a representative experiment are shown. See Supplementary Movie S3 for entire video sequence. Yellow arrows indicate broad lamellipodia-like protrusions frequently observed in TGFβ-treated cells in presence of Y27632. Scale bar, 100 μm. (B) Quantification of disseminating cells. The number of TGFβ-treated cells per field that in 2 days reached a distance >130 μm from the starting monolayer were counted, in absence or in presence of 10 μM Y27632, from three experiments (3 to 8 fields counted per experiment). (C) Rac1 activity is necessary for TGFβ-induced dissemination upon ROCK inhibition. 10 μM Y27632 ROCK inhibitor and 50 μM NCS237666 Rac1 inhibitor were added to TGFβ-treated cells as indicated. Number of cells TGFβ-treated n = 163, TGFβ/Y27632 treated n = 134, TGFβ/Y27632/NCS237666 treated n = 47, TGFβ/NCS237666 treated n = 42 from at least two experiments. (D) ROCK inhibition increases Rac1 activity at cellular protrusions. TGFβ-treated cells were transfected with a plasmid expressing Raichu-Rac1 (KRasCter) biosensor, treated with or without 10 μM Y27632 and visualized live by FRET microscopy the day after. Representative cells are shown. Scale bar 20 μm. (E) Quantification of whole-cell Rac1 activity. Number of untreated cells n = 54, Y27632 treated cells n = 33, from two experiments. Error bars represent SEM. p values come from two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
We reasoned that, in TGFβ-treated cells with high level of RhoA activity (Fig. 2C,D), the inhibition of Rho pathway leads to the up-regulation of Rac1 activity that may compensate the inhibition of contractility, presumably switching to a lamellipodia-driven invasion mechanism. Consistently, in TGFβ-treated cells, simultaneous inhibition of Rac1 by NSC23766 and of Rho pathway by Y27632 decreased dissemination speed (Fig. 6C, control versus Y27632/NSC23766); noteworthy, NSC23766 alone (i.e. in absence of Y27632) did not significantly impair the TGFβ-induced dissemination (Fig. 6C, control versus NSC23766), confirming that in this situation Rac1 GTPase is not at work.
These results indicate the existence of a complex balance and cross-talk between RhoA and Rac1 pathway in TGFβ-treated cells, as previously found in other cell models. In addition, these findings suggest that cancer cells, upon TGFβ-induced EMT, display a remarkable signaling plasticity which may contribute to adapt the motility/invasion programs to the chemical and physical properties of their microenvironment.
Discussion
Tumor cells can adopt various programs to move and invade, according to their genetic background and to biochemical and physical clues from their environment. The best characterized invasion mode is linked to proteolysis: cells secrete or locally accumulate matrix metalloproteinases (MMPs) that degrade the extra-cellular matrix (ECM) and create spaces through which they can advance. This proteolysis-dependent invasion program has been widely investigated also because it is easily recapitulated ex vivo, for example in Transwell invasion assays. However, there is a second major mode of invasion, based on deformation of the ECM by mechanical forces, that appears to be highly relevant in vivo39. A better characterization of force-dependent invasion is recently becoming possible thanks to the development of appropriate mechanobiological approaches, such as the Traction Force Microscopy used in this study.
We found that A549 lung cancer cells after undergoing an EMT display an increased capacity to generate forces and rely on a force-driven mode of locomotion to invade ECM. This promotes cell dissemination in a 2-3D invasion assay in which cells adhere to a glass surface at the ventral side, while their dorsal and front sides face Matrigel, a gelatinous protein mixture that resembles the basement membrane. Noteworthy, this setting mimics a situation of invasion at interfaces that is not rare in vivo. Indeed, recent advances in intravital imaging revealed that cancer cell invasion occurs preferentially via tissue tracks of least resistance, for example along vessels, myofibers and nerves40. Our results suggest that in the tumor microenvironment a local increase of TGFβ may contribute to cancer invasion and dissemination by a mechanism which relies on forces and contractility, without the need of proteolysis. We speculate that cancer cells, in particular after an EMT, may exploit this strategy to efficiently deform the pre-existing tracks and interstitial spaces surrounding tumors. The question that remains to be answered is whether the capacity to generate traction forces is a consequence of TGFβ stimulation, or of the resultant EMT, or of a combination of both. These two components cannot be easily separated.
If proteolysis of ECM is not always essential for invasion, why post-EMT cancer cells secrete great amounts of MMPs (Fig. 1F)? One possibility is that MMPs are required to do more than simply degrading physical barriers, as indicated by the emerging literature on their various regulating functions in the tumor microenvironment, including for example modulation of growth/survival, inflammatory response and angiogenesis41, functions that remain essential also during proteolysis-independent invasion.
It was previously shown that RalB controls migration, invasion and metastasis. Here we showed that, in a deformation-based mode of invasion, RalB controls contractility and force generation, via the association of its effector Exocyst with the RhoGEF GEF-H1 that activates the Rho-ROCK-MLC2 pathway (Fig. 7). Among the three Rho GTPases (RhoA, RhoaB, RhoC) activated by GEF-H142, we propose RhoA as the Rho player in the Ral-Rho cross-talk because the RhoA-containing biosensor monitored an increase of RhoA activating signals in TGFβ-treated A549 cells (Fig. 2C,D) and because RhoA is a well-established master regulator of cell contractility. However, a study with another cell model, the human colon LIM1863 cell line, reported a specific up-regulation of RhoC activity upon TGFβ stimulation43, leaving open the possibility of RhoC implication.

Model outlining the role of RalB/Exocyst pathway in the dissemination of TGFβ-treated A549 cells.
TGFβ-treated cells generate traction forces which are required to remodel the extracellular matrix and to disseminate in 2/3D environment. This requires the activation of the Rho/ROCK pathway and actomyosin function. The Ral pathway controls the dissemination of TGFβ-treated cells by modulating RhoA-dependent actomyosin contractility via the interaction between the Sec5 subunit of Exocyst and the RhoGEF GEF-H1.
In clinical trials protease inhibitors failed to prove any efficacy44,45. There is therefore urgency to search for alternative strategy to block cancer invasion. Our work suggests that inhibition of the activity of RalB or GEF-H1, by decreasing contractility-driven dissemination, could have a therapeutic anti-metastatic benefit for cancer patients. On the contrary, interfering more downstream by using ROCK inhibitors may promote worse outcome, because of activation of other motility machineries (Fig. 6). Deeper investigations are needed to validate or not these predictions; in particular, concerning GEF-H1, several studies are in agreement with a pro-migratory role30,31,32,33,34,35,36, but others suggest an anti-invasion role46, depending on the motility program33. In addition, GEF-H1 has also been shown to be required for oncogenic MAPK signaling, independently of its RhoGEF activity47, further increasing the potential interest of targeting this protein in anti-cancer drug discovery. On the RalB front, very recently, small molecules inhibitors for Ral proteins have been identified48, which opens the path for experimentally testing in the future the effects of anti-Ral therapy in vivo, both in animal models and in the clinic.
Globally, these findings provide a novel mechanistic insight on how RalB contributes to force-driven cancer invasion and dissemination and suggest that RalB function may be particularly crucial after EMT. Very interestingly, choices in motility modes are not irreversible: if the Rho-based engine is perturbed, cells might opt back for Rac-based motility (Fig. 6), so strong is the cancer eagerness of movements.
Materials and Methods
Cell culture
A549 lung adenocarcinoma cells were cultivated in RPMI 1640 GlutaMAX (#61870-010, Invitrogen) medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (Invitrogen, 15070-063), 1mM sodium pyruvate (#15070-063, Invitrogen) and glucose (#G8769, Sigma, 2.8 mL in 500 mL) at 37 °C in a humidified incubator with 5% CO2. Recombinant TGFβ1 (#240-B, R&D Systems) was added to the culture medium at 2 ng/ml concentration and refreshed every 2–3 days.
Plasmids
Raichu-RhoA 1218X and Raichu-Rac1 1011X biosensors were kindly provided by Michiyuki Matsuda (Kyoto University). To generate pLVX- Cherry-GEF-H1aa119–236 the sequence encoding Sec5 binding region of GEF-H1 (amino acids 119–236) was cloned as an EcoRI/BamHI fragment into pLVX lentiviral vectors (Clontech) containing mCherry (gift of Jasmine Abella).
DNA/siRNA transfections
For DNA transfections we used JetPRIME (#POL114, Polyplus) and for siRNA transfections Lipofectamine RNAiMax (#13778030, Invitrogen) according to manufacturer’s instructions. siRNAs were purchased from Eurogentec or Dharmacon, see Supplementary Materials and Methods for sequences.
Viral production and infection
Stable A549 cell lines expressing mCherry alone or Cherry-GEF-H1aa119–236 were generated via infection using lentiviruses. For viral production, 293T cells were transfected by Lipofectamine 2000 (#11668027, Invitrogen) with pLVX viral vectors along with lentiviral packaging plasmid (psPAX2) and VSVG expression vector (pCMV-VSV-G). Viral supernatant was harvested at 72 hrs post-transfection, filtered and added to the recipient cell lines with 6 μg/ml polybrene (# 107689, Sigma-Aldrich) for 24 hrs infection. Infected cells were then selected with 2 μg/ml puromycin (# ant-pr, Invivogen).
Circular Invasion Assay
Circular Invasion Assay (CIA) was performed as previously described20. Briefly, before seeding 1 × 106 A549 cells a silicon self-stick cellular stopper (#80209, Ibidi) was placed in the center of a 35-mm uncoated glass bottom dish (#8158, Ibidi) to create a square space (0.80 cm2) devoid of cells. Upon cell adhesion, the stopper is removed and 300 μl of 50% GFR-Matrigel (#354230, Corning) (4.5 mg/ml) in 1X PBS was overlaid onto the cell monolayer seeded in the inner circle of the dish to create a matrix gel (about 0.8 mm high) which covers the dorsal part of the cells and fills the empty square space. The gel was allowed to polymerize for 2 hrs prior to addition of growth medium. Images of invading cells were acquired every 15 or 60 min using a Leica DMIR2 inverted microscope equipped with 5X or 10X objective, motorized stage, Coolsnap HQ2 camera (Roper Scientific), “Box” heated chamber and “Brick” CO2 controller (Life Imaging Services), under the control of MetaMorph software (Universal Imaging). The Manual Tracking plug-in (developed by Fabrice Cordelier) of ImageJ software was used to track cells, after which data were exported to Excel for mathematical and statistical analysis.
FRET Imaging
Cells expressing Raichu biosensor were grown and imaged on 35-mm uncoated glass bottom dish (#8158, Ibidi). 24 hrs after transfection cells were imaged in phenol red-free MEM medium (#51200-038, Invitrogen) containing 2% FBS using a Leica DMIRE2 inverted microscope, equipped with an HCX PL APO 63× oil immersion objective, CoolSNAP HQ2 camera (Roper Scientific), filter wheels (Ludl Electronic Products), “Box” heated chamber and “Brick” CO2 controller (Life Imaging Services), under the control of MetaMorph software (Universal Imaging). For dual-emission ratio imaging, we used a D440/20× excitation filter, a 455DCLP dichroic mirror and the two D485/40m (CFP) and D535/30m (YFP) emission filters (Chroma, Filter Set #71007a). Cells were illuminated with a 103-watt mercury lamp (Osram) through a 5% transmission neutral density (ND) filter. The exposure time was 0.05–0.2 s with a camera binning of 4 × 4. Image processing was performed using MetaMorph. For whole cell FRET measurements, the background-subtracted images were thresholded to define the whole cell surface, then the mean cell fluorescence intensities (CFP, YFP) were measured and the values were exported to Excel spreadsheets for mathematical treatment and statistics calculations. For FRET visualization, background-subtracted CFP images were thresholded to create a binary mask with a value of 0 outside the cell and a value of 1 inside the cell; after multiplication of CFP and YFP images by this mask, the FRET (YFP/CFP ratio) is represented using an eight-color scale code, as shown on the right of time-lapse figures with the upper and lower limits; the YFP/CFP ratio is used so that high ratio (red) corresponds to high Rho/Rac activity and low ratio (blue) to low Rho/Rac activity.
Contraction assay
To assess force-mediated collagen contraction 2 × 105 A549 cells were embedded in 200 μL of a collagen solution and seeded on a 35-mm glass-bottom MatTek dish (P35-1.5-14-C, MatTek). Collagen solution (1 mL) was prepared by mixing 0.8 mL of PureCol Bovine Collagen I (#5005-B, Advanced BioMatrix), 0.2 mL of 5X DMEM medium (D2429, Sigma) and 40 μL of 7.5% NaHCO3 pH = 8. After polymerization, gels with cells were maintained in culture media. Gel contraction was quantified by taking photographs (iPhone 4S) after 4 days. The relative diameters of the well and the gel were measured using ImageJ software and the percentage of contraction was calculated using the formula: 100 x (well diameter-gel diameter)/well diameter.
Traction Force Microscopy
Acrylamide gels containing fluorescent beads were prepared following previously published protocols49. The concentrations of acrylamide (polymer) and bis-acrylamide (cross-linker) were adapted to get a final Young modulus of 10.6 kPa49. Briefly, we placed a drop of the mixture of acrylamide/bis-acrylamide between a fibronectin-coated glass coverslip (coated with 10 mg/mL fibronectin in PBS (Fibronectin Bovine Protein, Plasma, Life technologies)) and an amino-silanated glass coverslip (silanised with 3-aminopropyltriethoxylsilane (Sigma) in isopropanol (1mL silane/50mL isopropanol)). The mixture was then crosslinked by UV light after which the top coverslip was carefully detached to obtain a gel slab attached to the silanized coverslip and coated with fibronectin. After incubating the gels for 1 hr in culture medium at 37 °C and 5% CO2, the slides were ready to be used. We seeded the cells at low density (~50 cells/mm2) to ensure that traction forces were measured on single cells. Gels were imaged on an inverted spinning disk confocal (Inverted Wide Confocal Spinning Disk Roper/Nikon) with a 40X oil lens. We took images of the relaxed gels after removing the cells with trypsin (Sigma). Images of the beads with and without the cells were first aligned using the ImageJ plugin “Align slices in stack”. We then used Particle Image Velocimetry (PIV) to measure the beads displacement field and the Fast Fourier Transform traction cytometry (FTTC) as previously described in50 in order to reconstruct the traction force field. The regularization parameter was set to 9 × 10−10 for all traction force reconstructions. The PIV and FFTC grid was set to 1.93 μm × 1.93 μm. Images were processed with the ImageJ software51. Further analysis was performed with Matlab (MathWorks, Natick, MA). To quantify cell contractility, we measured the strain energy Estrain which is the total energy transfered from the cell to the elastic distortion of the substrate24.

 is the traction field from the FFTC and
is the traction field from the FFTC and  is the displacement field from the PIV measurement.
is the displacement field from the PIV measurement.
Immunofluorescence, Immunoblotting, zymography and invadopodia assay
See Supplementary Materials and Methods.
Additional Information
How to cite this article: Biondini, M. et al. RalB regulates contractility-driven cancer dissemination upon TGFβ stimulation via the RhoGEF GEF-H1. Sci. Rep. 5, 11759; doi: 10.1038/srep11759 (2015).
References
Bodemann, B. O. & White, M. A. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer 8, 133–140 (2008).
Gentry, L. R., Martin, T. D., Reiner, D. J. & Der, C. J. Ral small GTPase signaling and oncogenesis: More than just 15 minutes of fame. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1843, 2976–2988 (2014).
Hamad, N. M. et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 16, 2045–2057 (2002).
Chien, Y. & White, M. A. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 4, 800–806 (2003).
Oxford, G. et al. RalA and RalB: antagonistic relatives in cancer cell migration. Cancer Res. 65, 7111–7120 (2005).
Rossé, C. et al. RalB mobilizes the exocyst to drive cell migration. Mol. Cell. Biol. 26, 727–734 (2006).
Lim, K.-H. et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. CB 16, 2385–2394 (2006).
Rybko, V. A. et al. Different metastasis promotive potency of small G-proteins RalA and RalB in in vivo hamster tumor model. Cancer Cell Int. 11, 22 (2011).
He, B. & Guo, W. The exocyst complex in polarized exocytosis. Curr. Opin. Cell Biol. 21, 537–542 (2009).
Parrini, M. C. et al. SH3BP1, an exocyst-associated RhoGAP, inactivates Rac1 at the front to drive cell motility. Mol. Cell 42, 650–661 (2011).
Friedl, P. & Alexander, S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 (2011).
Charras, G. & Sahai, E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 15, 813–824 (2014).
Wolf, K. & Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 21, 736–744 (2011).
Massagué, J. TGFbeta in Cancer. Cell 134, 215–230 (2008).
Pickup, M., Novitskiy, S. & Moses, H. L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 13, 788–799 (2013).
Lamouille, S., Xu, J. & Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014).
Tsai, J. H. & Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 27, 2192–2206 (2013).
Kasai, H., Allen, J. T., Mason, R. M., Kamimura, T. & Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 6, 56 (2005).
Tang, Y., Shu, G., Yuan, X., Jing, N. & Song, J. FOXA2 functions as a suppressor of tumor metastasis by inhibition of epithelial-to-mesenchymal transition in human lung cancers. Cell Res. 21, 316–326 (2011).
Yu, X. & Machesky, L. M. Cells assemble invadopodia-like structures and invade into matrigel in a matrix metalloprotease dependent manner in the circular invasion assay. PloS One 7, e30605 (2012).
Weigelin, B., Bakker, G.-J. & Friedl, P. Intravital third harmonic generation microscopy of collective melanoma cell invasion. IntraVital 1, 32–43 (2012).
Eckert, M. A. et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19, 372–386 (2011).
Saito, R.-A. et al. Thyroid transcription factor-1 inhibits transforming growth factor-beta-mediated epithelial-to-mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 69, 2783–2791 (2009).
Butler, J. P., Tolić-Nørrelykke, I. M., Fabry, B. & Fredberg, J. J. Traction fields, moments and strain energy that cells exert on their surroundings. Am. J. Physiol. Cell Physiol. 282, C595–605 (2002).
Olson, M. F. & Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 26, 273–287 (2009).
Pathak, R. et al. The microtubule-associated Rho activating factor GEF-H1 interacts with exocyst complex to regulate vesicle traffic. Dev. Cell 23, 397–411 (2012).
Birkenfeld, J. et al. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 12, 699–712 (2007).
Guilluy, C. et al. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 13, 722–727 (2011).
Heck, J. N. et al. Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol. Biol. Cell 23, 2583–2592 (2012).
Tsapara, A. et al. The RhoA Activator GEF-H1/Lfc Is a Transforming Growth Factor-β Target Gene and Effector That Regulates α-Smooth Muscle Actin Expression and Cell Migration. Mol. Biol. Cell 21, 860–870 (2010).
Liao, Y. C. et al. Overexpressed hPTTG1 promotes breast cancer cell invasion and metastasis by regulating GEF-H1/RhoA signalling. Oncogene 31, 3086–3097 (2012).
Ridgway, L. D., Wetzel, M. D., Ngo, J. A., Erdreich-Epstein, A. & Marchetti, D. Heparanase-induced GEF-H1 signaling regulates the cytoskeletal dynamics of brain metastatic breast cancer cells. Mol. Cancer Res. MCR 10, 689–702 (2012).
Von Thun, A. et al. Extracellular signal-regulated kinase regulates RhoA activation and tumor cell plasticity by inhibiting guanine exchange factor H1 activity. Mol. Cell. Biol. 33, 4526–4537 (2013).
Cheng, I. K. C. et al. GEF-H1 over-expression in hepatocellular carcinoma promotes cell motility via activation of RhoA signalling. J. Pathol. 228, 575–585 (2012).
Nalbant, P., Chang, Y.-C., Birkenfeld, J., Chang, Z.-F. & Bokoch, G. M. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol. Biol. Cell 20, 4070–4082 (2009).
Eitaki, M., Yamamori, T., Meike, S., Yasui, H. & Inanami, O. Vincristine enhances amoeboid-like motility via GEF-H1/RhoA/ROCK/Myosin light chain signaling in MKN45 cells. BMC Cancer 12, 469 (2012).
Vega, F. M., Fruhwirth, G., Ng, T. & Ridley, A. J. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 193, 655–665 (2011).
Sanz-Moreno, V. et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523 (2008).
Wyckoff, J. B., Pinner, S. E., Gschmeissner, S., Condeelis, J. S. & Sahai, E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. CB 16, 1515–1523 (2006).
Alexander, S., Weigelin, B., Winkler, F. & Friedl, P. Preclinical intravital microscopy of the tumour-stroma interface: invasion, metastasis and therapy response. Curr. Opin. Cell Biol. 25, 659–671 (2013).
Kessenbrock, K., Plaks, V. & Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010).
Ren, Y., Li, R., Zheng, Y. & Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 273, 34954–34960 (1998).
Bellovin, D. I. et al. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene 25, 6959–6967 (2006).
Coussens, L. M., Fingleton, B. & Matrisian, L. M. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 (2002).
Fingleton, B. MMPs as therapeutic targets--still a viable option? Semin. Cell Dev. Biol. 19, 61–68 (2008).
Osborne, L. D. et al. TGF-β regulates LARG and GEF-H1 during EMT to impact stiffening response to force and cell invasion. Mol. Biol. Cell (2014). 10.1091/mbc.E14-05-1015
Cullis, J. et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell 25, 181–195 (2014).
Yan, C. et al. Discovery and characterization of small molecules that target the GTPase Ral. Nature 515, 443–447 (2014).
Tse, J. R. & Engler, A. J. Preparation of hydrogel substrates with tunable mechanical properties. Curr. Protoc. Cell Biol. Editor. Board Juan Bonifacino Al Chapter 10, Unit 10.16 (2010).
Tseng, Q. et al. Spatial organization of the extracellular matrix regulates cell-cell junction positioning. Proc. Natl. Acad. Sci. U. S. A. 109, 1506–1511 (2012).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
We thank Sarah Moitrier, Timo Betz and Gregory Beaune for helpful discussion on the TFM measurement, Nathalie Brandon for technical assistance in molecular biology, Anthony Simon for tests with spheroids, Pedro Monteiro, Antonio Castro, Catalina Lodillinsky from Philippe Chavrier laboratory for kindly sharing their expertise on invadopodia and metalloproteinases. Some experiments were performed on workstations of the PICT-IBiSA Lhomond Imaging facility of Institut Curie, in particular with the help of Olivier Renaud and Olivier Leroy for light microscopy. MB was recipient of fellowships of Ligue National contre le Cancer and Fondation ARC pour la Recherche sur le Cancer and NM-S was supported by a MHV grant from the Swiss National Science Foundation. This work was supported by the Fondation ARC pour la Recherche sur le Cancer (MCP SFI20121205710, JC SFI20111203931 and SFI20121205686), Ligue National contre le Cancer (MCP RS14/75-54, PS Equipe labellisée), Agence Nationale de la Recherche (ANR-11 BSV2 012 01), the European Research Council (F. Del Bene, ERC ZEBRATECTUM N°311159), the Fondation Pierre-Gillles de Gennes pour la recherche (JC, PS) and Association Christelle Bouillot (JC).
Author information
Authors and Affiliations
Contributions
M.C.P. and J.C. conceived the project. M.B., P.S., M.C.P. designed the experiments. M.B., G.D., N.M.-S. performed and analyzed the experiments. M.B. and M.C.P. wrote the manuscript. All the authors commented on the manuscript and approved its submission.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Biondini, M., Duclos, G., Meyer-Schaller, N. et al. RalB regulates contractility-driven cancer dissemination upon TGFβ stimulation via the RhoGEF GEF-H1. Sci Rep 5, 11759 (2015). https://doi.org/10.1038/srep11759
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11759
This article is cited by
-
The role of ral signaling and post translational modifications (PTMs) of Ras in cancer
Genome Instability & Disease (2022)
-
Localization of RalB signaling at endomembrane compartments and its modulation by autophagy
Scientific Reports (2019)
-
Integrin-Dependent Regulation of Small GTPases: Role in Cell Migration
Journal of the Indian Institute of Science (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
