
Abstract
Plant virus coat proteins (CPs) play a fundamental role in protection of genomic RNAs, virion assembly and viral movement. Although phosphorylation of several CPs during virus infection have been reported, little information is available about CP phosphorylation of the spherical RNA plant viruses. Here, we demonstrate that the CP of Beet black scorch virus (BBSV), a member of the genus Necrovirus, can be phosphorylated at threonine-41 (T41) by cAMP-dependent protein kinase (PKA)-like kinase in vivo and in vitro. Mutant viruses containing a T41A non-phosphorylatable alanine substitution and a T41E glutamic acid substitution to mimic threonine phosphorylation were able to replicate but were unable to move systemically in Nicotiana benthamiana. Interestingly, the T41A and T41E mutants generated unstable 17 nm virus-like particles that failed to package viral genomic (g) RNA, compared with wild-type BBSV with 30 nm virions during viral infection in N. benthamiana. Further analyses showed that the T41 mutations had little effect on the gRNA-binding activity of the CP. Therefore, we propose a model whereby CP phosphorylation plays an essential role in long-distance movement of BBSV that involves formation of stable virions.
Similar content being viewed by others


Introduction
Beet black scorch virus (BBSV) belongs to the genus Necrovirus in the family Tombusviridae and was first reported in the late 1980s in China1, followed by identification in the US, Iran and Spain2,3,4,5,6. Under natural conditions, BBSV-infected beets have scorched and inwardly-curled leaves and necrotic roots, which result in severe reductions in sugar production7. BBSV transmission in the soil is facilitated by zoospores of Olpidium brassicae8 and at least 15 plants from 4 families, including Chenopodium amaranticolor, Tetragonia expanse and Nicotiana benthamiana, can be mechanically infected by BBSV7,9,10.
BBSV is a small, approximately 30 nm icosahedral virus, consisting of a 3644 nucleotide (nt) plus-sense, single-stranded (ss) genomic (g) RNA lacking a 5’-cap structure and a 3’-poly(A) tail that encodes six functional proteins. The gRNA serves as the mRNA for the 5’ proximal ORF, which encodes a 23 kDa protein (p23) and an 82 kDa readthrough protein (p82). P23 and p82 interact to form the replicase complex, which is required for gRNA replication and synthesis of two subgenomic RNAs (sgRNAs). Each of the three proteins (p7a, p7b and p5’) translated from sgRNA1 are required for viral cell-to-cell movement and the 24.5 kDa coat protein (CP), which is translated from sgRNA2, is required for systemic infection of N. benthamiana, but is dispensable for cell-to-cell movement in C. amaranticolor11,12,13.
The CPs of the Tombusviridae are composed of an RNA binding domain (R-domain), a shell domain (S-domain), a hinge domain (H-domain) and a protruding domain (P-domain)14,15. Tombusvirus CPs are also multifunctional and have many important roles during the infection cycle, including nucleic acid binding and encapsidation16,17,18,19. CP binding specificity for the gRNA is critical for initiation of virion assembly and is R-domain dependent20,21. In the case of BBSV, recent studies have demonstrated that an N-terminal region enriched in basic amino acids (4KRNKGGKKSR13) is involved in CP nuclear localization and gRNA binding and that lysine and arginine at positions 4 and 5 are key amino acid requirements for gRNA binding16,22. Although not critical for direct interactions with gRNA, other residues are also required for virion assembly16. Mutations of these residues impact virion assembly and viral systemic movement to different degrees and disruption of these functions supports a model positing that virions are essential for necrovirus systemic movement16,23.
A number of studies have shown that CP stability and functions of diverse plant viruses, such as Cauliflower mosaic virus (CaMV), Potato virus A (PVA), Plum pox virus (PPV), Bamboo mosaic virus (BaMV), Potato virus X (PVX), Groundnut bud necrosis virus (GBNV) and Brome mosaic virus (BMV), are regulated by phosphorylation24,25,26,27,28,29,30,31,32. Phosphorylation of the CaMV precapsid protein (p57) by casein kinase II (CKII) activates proteolytic processing to form a “mature” N and C-terminal-truncated p37 product that is essential for successful infection. CP phosphorylation also regulates translation of the gRNAs of several filamentous plant RNA viruses. PVX encapsidated gRNA is completely non-translatable in vitro, but when the N-terminal CP residues are phosphorylated by protein kinase C (PKC) or by a casein kinase mixture (CKI and CKII), the RNA is converted to a translatable form26. Another case involves the PVA and BaMV CP, in which phosphorylation at the RNA binding domain reduces RNA binding affinity. Both Hung et al. (2014) and Ivanov et al. (2001 and 2003) hypothesize that CP phosphorylation promotes virion or ribonucleoprotein (RNP) disassembly to release gRNA for replication or translation27,28,33. Hence, several lines of evidence with potexviruses and potyviruses suggest that CP phosphorylation is a general process that regulates co-translational disassembly of virions or RNP and viral cell-to-cell movement27,28,33.
Although several models have suggested that CP phosphorylation modulates plant virus or RNP disassembly after cell entry, regulation of virion assembly by phosphorylation remains elusive. Weiland et al. (2007) have reported that BBSV virions can be phosphorylated3, however, more detailed analyses have not been described. In the present study, we provide evidence that the BBSV CP is phosphorylated during infection of N. benthamiana. We have also conducted comprehensive biochemical approaches, including mass spectrometry and phosphorylation assays that demonstrate that the CP is phosphorylated at threonine 41 (T41) in vivo and in vitro by PKA-like kinases. Our results also indicate that CP T41 mutants form incompletely-assembled virus-like particles (VLPs) that are unable to package gRNA and are less stable than wild-type (wt) virions and that these mutants compromised in systemic movement during infection of N. benthamiana. In summary, our results reveal a correlation between CP phosphorylation, virion assembly and long-distance movement, which has been largely unexplored for spherical RNA plant viruses.
Results
Identification of phosphorylated residues that affect normal CP functions during BBSV infection
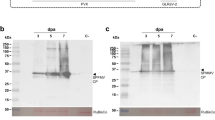
To determine whether the BBSV CP is subjected to phosphorylation during virus infection, CP immunoprecipitated from BBSV-infected N. benthamiana leaf tissues was separated by SDS-PAGE (Fig. 1a). After in-gel digestion with trypsin, the CP was analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) and four potential phosphorylation sites, S12, S15, T18 and T41, were identified (Fig. 1b,c). To determine whether CP phosphorylation affects BBSV infection, one or several amino acid residues were substituted with alanines (A) and introduced into the BBSV infectious cDNA clone, pUBF52 (Supplementary Fig. S1). Two-week-old N. benthamiana plants were inoculated with BBSVwt or BBSV mutant in vitro transcripts and incubated in a plant growth chamber at 18 °C for about 3 weeks. During early infections, BBSV mutants elicited a local chlorosis on the inoculated leaves similar to that of BBSVwt (Fig. 2a). To detect CP accumulation, leaf tissues were harvested at 14 days post-inoculation (dpi) and subjected to enzyme-linked immunosorbent assays (ELISA, Fig. 2b). CP accumulation of the BBSVT41A (Fig. 2b, lane 6) and BBSVS12/S15/T18/T41A mutants (Fig. 2b, lane 8) was reduced dramatically in comparison to that of BBSVwt (Fig. 2b, lane 2), whereas CP accumulation of BBSVS12A, BBSVS15A, BBSVT18A and BBSVS12/S15/T18A was similar to that of BBSVwt (Fig. 2b, lanes 3–5 and 7). Viral replication and CP expression were also analyzed by Northern and Western blot, respectively and consistent results were obtained (Fig. 2c,d). Plants inoculated with the T41A mutants did not developed visible chlorotic spots by 21 dpi, (Fig. 2e, lanes 6 and 8) and ELISA, Northern and Western blot assays did not reveal detectable virus accumulation (Fig. 2f–h, lanes 6 and 8). However, leaves inoculated with other BBSV mutants (Fig. 2e–h, lanes 3–5 and 7) exhibited systemic symptoms and accumulated virus levels similar to those of BBSVwt (Fig. 2e–h, lane 2). These results indicate that phosphorylation of CP T41, but not the other candidate phosphorylation sites, is required for systemic infection of BBSV in N. benthamiana.

Identification of phosphorylation sites of CP by LC-MS/MS.
(a) Coomassie blue staining of CP immunoprecipitated (IP) from mock or BBSV inoculated N. benthamiana leaf tissues. The CP band (arrowhead) was digested in gel and then subjected to LC-MS/MS analysis. (b) Mass spectral sequence coverage (amino acids presented with underline) and identified phosphorylation sites (amino acids presented in bold) in the BBSV CP. (c) MS2 spectrum of the doubly charged phosphopeptide 38IRSpTTIGTR46 (m/z 542.78) containing the T41 phosphorylated residue. The colored highlights show b and y ions in the sequence and the corresponding peaks in the spectrum.

Accumulation of BBSVwt and phosphorylation site mutant viruses in N. benthanmiana.
N. benthamiana leaves exhibiting local (a) and systemic (e) symptoms after inoculation with BBSVwt and BBSV mutants containing non-phosphorylatable CP substitutions. ELISA detection with CP-specific antibodies of leaf extracts was conducted on inoculated leaves at 14 dpi (b) and upper leaves at 21 dpi (f). CP titers calculated from A405 ELISA values are indicated on the Y-axis. Viral gRNA replication and CP expression were analyzed in inoculated and upper leaves by Northern blot (c and g) and Western blot (d and h), respectively. The number ratios of symptom induction among the total inoculated plants are showed at the bottom of panels a and e.
CP phosphorylation is associated with PKA-like kinases
According to a previous report34 describing phosphorylation specificity of proteins and prediction results from GPS 2.135 (http://gps.biocuckoo.org/index.php) and the online servers KinasePhos36 (http://KinasePhos2.mbc.nctu.edu.tw/), the 38IRST41 region in the N-terminus of BBSV CP is similar to the R-R-X-[S/T] motif, which is a conserved substrate recognition site of cAMP-dependent protein kinase (PKA). To determine whether BBSV CP is phosphorylated by PKA, total protein was extracted from BBSV-infected-N. benthamiana leaf tissues at 14 dpi and subjected to Western blot with a phospho-(Ser/Thr) PKA substrate antibody (α-pS/T) and a CP-specific antibody (α-CP). A weak phosphorylated band corresponding to the CP position was present in the crude extracts from BBSV-infected plants (Fig. 3a, lane 2, top panel), but not from mock-inoculated plant extracts (Fig. 3a, lane 1, top panel). After CP enrichment by CP-specific polyclonal antibody and co-immunoprecipitation with protein G-beads, the phosphorylated CP band displayed higher intensity (Fig. 3a, lane 4, top panel). In addition, phosphorylation of the BBSV CPT41A was approximate 60% of BBSV CPwt immunoprecipitated from inoculated plants (Fig. 3a, lane 5, top panel). These results provided preliminary evidence that the BBSV CP is phosphorylated at T41 by PKA-like kinases in N. benthamiana.

Phosphorylation of BBSV CP by PKA in vivo and in vitro.
(a) Detection of phosphorylation of CP by PKA in N. benthamiana. Total protein was extracted from BBSVwt-inoculated leaves and subjected to Western blots using phospho-(Ser/Thr) PKA substrate antibodies (α-pS/T, top panel, lane 2) or CP-specific antibodies (α-CP, bottom panel, lane 2). Then total extracted proteins from BBSVwt and BBSVT41A inoculated leaves were also immunoprecipitated with CP-specific antibodies and the same volumes of recovered CPwt and CPT41A were analyzed with CP antibodies (bottom panel, lanes 4 and 5). For the phospho-(Ser/Thr) PKA antibody tests, gels were loaded with three times the amounts used for the CP antibody analyses (top panel, lanes 4 and 5). The mobility of the CP band is indicated by arrowheads and mock-inoculated N. benthamiana extracts served as negative controls (lanes 1 and 3). (b) Coomassie blue staining of His-tagged recombined CP (rCP, arrowhead) from uninduced (lane 1) or IPTG induced (lane 2) E. coli. The insoluble (lane 3) and soluble (lane 4) rCPs were separated by centrifugation and the soluble fraction was purified by Ni-NTA affinity chromatography (lanes 4–8) and analyzed by SDS-PAGE. Molecular size markers are indicated on the left and the rCP is indicated on the right by an arrowhead. (c) In vitro phosphorylation of rCP by purified CKII (lane 3), commercial CaMKII (lane 6) and commercial PKA (lane 9, arrowhead). Reactions containing GST-tagged kinase specific substrates served as positive controls (lanes 4, 7 and 10) and reactions lacking rCP (lanes 2, 5 and 8) or specific kinases (lane 1) served as negative controls. The autoradiography (top) and Coomassie blue stained loading controls (bottom) are shown. (d) Validation of in vitro rCP phosphorylation with the H-89 PKA specific inhibitor. Samples were treated with buffers lacking (lane 3) or containing 1 μM (lane 4) and 10 μM (lane 5) of H-89. Autoradiography and equal loading are shown in the top and bottom panels, respectively. (e) In vitro phosphorylation assays indicated that T41 is a PKA substrate. His-tagged CPwt, CPT41A, CPT41E and CPR39A were phosphorylated in vitro by PKA and autoradiographed (top panel, arrowhead) and loading controls are indicated at the bottom panel.
To provide additional evidence to confirm that CP is a substrate for PKA and that T41 is the phosphorylation target site, we performed a series of in vitro phosphorylation assays. First, recombinant CP (rCP) fused with an N-terminal hexahistidine tag was expressed in E. coli and affinity purified over Ni-NTA agarose (Fig. 3b) and used for in vitro phosphorylation assays with commercial PKA (New England Biolabs). As kinase specificity controls, we also used the recombinant N. benthamiana CKII (NbCKII) purified as previously described33 and a commercial calcium/calmodulin-dependent protein kinase II (CaMKII, New England Biolabs). These experiments revealed that the BBSV CP is specifically phosphorylated by PKA (Fig. 3c, lane 9), but not by NbCKII (Fig. 3c, lane 3) or CaMKII (Fig. 3c, lane 6). To provide additional evidence regarding PKA phosphorylation specificity, the H-89 PKA inhibitor37 was added to the reactions (Fig. 3d). In the absence of H-89, an intense radioactive CP band was evident (Fig. 3d, lane 3), but radioactivity was reduced substantially in the presence of 1 μM H-89 (Fig. 3d, lane 4) and was not evident when the H-89 concentration was increased to 10 μM (Fig. 3d, lane 5). Thus the results in total, demonstrate that BBSV CP is a PKA substrate in vitro.
In additional experiments, a non-phosphorylatable T41A mutant, a phosphorylation mimicking T41E mutant and an R39A mutant designed to interfere with the PKA phosphorylation consensus motif R-R-X-[S/T]34 were constructed and compared with in vitro PKA phosphorylation of rCPwt. Three independent experiments consistently revealed that rCPT41A, rCPR39A and rCPT41E displayed approximately 50, 67 and 52% phosphorylation efficiency compared with CPwt (Fig. 3e, lanes 3–6). Based on these results, we conclude that CP T41 is phosphorylated in vivo and in vitro by PKA.
CP T41 phosphorylation impacts BBSV long-distance movement but not replication
To determine whether phosphorylation of CP T41 affects BBSV movement, we compared the symptom induction and accumulation of BBSVwt virions with those of BBSVT41A, BBSVT41E and BBSVR39A mutants. Consistent with BBSVT41A, which can not be phosphorylated (Fig. 4e–h, lane 3), the BBSVT41E mutant that most closely resembles phosphorylated threonine, did not elicit discernible systemic symptoms (Fig. 4e, lane 4) and did not accumulate detectable CP and viral RNA (Fig. 4f–h, lane 4). These results suggest that interference with T41 phosphorylation impairs systemic infection. Upper leaves of plants inoculated with BBSVR39A developed systemic symptoms, but the proportion of leaves developing symptoms was lower (Fig. 4e, lane 5) and the viral accumulation and infection rates (Fig. 4, lane 5) were also lower than those of BBSVwt (Fig. 4, lane 2). Taken together, these results indicate that phosphorylation of CP at T41 contributes to BBSV long-distance movement in N. benthamiana.

Accumulation of BBSVwt and mutant viruses in N. benthamiana.
N. benthamiana leaves exhibiting local (a) and systemic (e) BBSVwt and mutant viruses symptoms at 14 and 21 dpi. CP accumulation was determined by ELISA (b and f) and Western blot (d and h). Replication of viral RNA in inoculated and upper leaves was examined by Northern blot (c and g). The infection rate is indicated at the bottom of panels a and e.
After determining that CP T41 phosphorylation is involved in BBSV systemic infection, but has little apparent impact on cell-to-cell movement, we were curious about whether CP phosphorylation might have more subtle effects on viral replication. N. benthamiana protoplasts were transfected with the BBSVwt or T41 mutants. After 20 hours post-inoculation (hpi), total RNA and protein were extracted from the infected protoplasts and subjected to Northern and Western blot analyses. The accumulation levels of the progeny RNAs (Fig. 5a) and CP (Fig. 5b) of the T41 mutants and BBSVwt-infected protoplasts were similar. Hence, phosphorylation of T41 appears not to have obvious effects on the initial accumulation of BBSV RNA or on expression of the BBSV CP. However, by 60 hpi, accumulation of the mutant RNAs was substantially lower than those of BBSVwt (Fig. 5c) and the accumulation levels of the mutant CPs were also lower than the CPwt (Fig. 5d). These results suggest that phosphorylation of T41 does not affect the initial stages of replication, but that viral accumulation is impaired as replication progresses, so it is also possible that CP phosphorylation is required for effective interference with host defense mechanisms at later stages of replication.

Detection of viral accumulation in N. benthamiana protoplasts transfected with BBSVwt and T41 mutant viruses.
Total RNA extracted from BBSVWT and T41 mutant inoculated protoplasts at 20 or 60 hpi was subjected to Northern blot with a DIG-labeled BBSV-specific probe (a and c. top panel). EtBr staining shows the rRNA loading controls (a and c, bottom panel) and gRNA and sgRNAs are indicated on the left. Total proteins extracted from the protoplasts were subjected to Western blot with CP-specific antibodies (b and d, top panel). Equal proteins loading is verified by Coomassie blue staining (b and d, bottom panel) and uninfected protoplasts (mock) served as a negative control.
T41 CP mutants are defective in virus assembly
It has been reported that stable virions are essential for BBSV systemic movement16. Therefore, to determine whether T41 and R39 mutant viruses can form intact virions during infection, BBSVwt, BBSVT41A, BBSVT41E and BBSVR39A-inoculated leaf samples were homogenized in PBS buffer, given a low speed centrifugation and the clarified supernatants were observed by transmission electron microscopy (TEM) (Fig. 6a, top panel). The mock-inoculated control did not contain visible virus-like particles (Fig. 6a, A), whereas BBSVwt and BBSVR39A-infected samples displayed readily observed particles with 30.09 ± 0.68 nm and 27.56 ± 1.62 nm in diameters (Fig 6a, B and E). In contrast, the BBSVT41A and BBSVT41E-infected leaf samples contained aberrant particles with diameters of 16.82 ± 1.57 nm and 16.97 ± 1.14 nm, respectively and these particles tended to form irregular aggregates (n = 50, Fig 6a, C and D). To evaluate the aberrant particles in more detail, partial purifications consisting of a sodium acetate buffer (pH 5) extraction and PEG precipitation were carried out for preliminary enrichment38 and the middle panel of Fig. 6a shows that these enriched particles were similar to those in the clarified leaf sap (Fig. 6a, top panel). However, further conventional and refined BBSVT41A and BBSVT41E purifications39 did not contain virus-like particles (Fig. 6a, bottom panel, M and N), in contrast to the 30 and 28 nm particles that were readily recovered from the refined BBSVwt and BBSVR39A preparations (Fig. 6a, bottom panel, L and O). We attribute these differences to dissociation of the aberrant particles during extraction in the neutral pH phosphate buffer, which does not contain stabilizing Ca2+ ions and to the subsequent harsh organic solvent clarification. Hence, these results suggest that the BBSVT41A or BBSVT41E mutant virus particles have defects in morphogenesis and are less stable than the BBSVwt particles and we propose that the systemic movement defects exhibited by T41 mutant viruses are a consequence of incomplete virion formation.

Analysis of particles from N. benthamiana leaves inoculated with BBSVwt and mutant viruses.
(a) Transmission electron microscopy (TEM) of VLPs obtained by partial and refined purification procedures from N. benthamiana plants inoculated with BBSVwt, BBSVT41A, BBSVT41E and BBSVR39A transcripts. Inoculated leaf tissue was harvested at 14 dpi, extracted and observed by TEM (top panel). The VLPs were further enriched by partial or refined purification procedures and detected by TEM (middle and bottom panel). Mock-inoculated leaf samples served as controls (A, F and K). Bars = 100 nm. More than 50 particles in each sample were measured and their average diameters are shown under the top panel. (b) VLP CP detection by Western blot with the CP antibodies used 5 μl samples from partial (middle panel) and refined (bottom panel) purifications. Total protein extracted from inoculated leaf tissues served as controls (top panel). (c) EtBr detection of viral RNA from partially purified BBSVwt and mutant preparations (2 μg) after 1% agarose gel electrophoresis (lanes 3–6). BBSVwt virions from the refined purifications served as a positive control to identify the encapsidated RNA (lane 1). Mock-inoculated leaf extracts subjected to the partial purification served as a negative control (lane 2).
Aberrant particles formed by T41 mutant viruses are deficient in RNA packaging
To investigate the composition of particles formed by BBSVwt and mutant viruses, particles obtained from the partial and refined purifications were subjected to Western blot analyses using CP-specific antibodies. The results verified that BBSVwt inoculations contained high amounts of CP, whereas the partially purified T41 and R39 mutant BBSV preparations contained lower amounts of CP (Fig. 6b, middle panel). Moreover, the lack of detectable T41 mutant CP in refined purifications strongly suggests that small aberrant virus-like particles are unstable. Interestingly, although BBSVR39A formed virus particles, the particle yield (0.5 mg per 10 g leaf tissue) was less than half the amount recovered from BBSVwt samples (1.35 mg per 10 g leaf tissue) after the refined purification (Fig. 6b, bottom panel), suggesting that the R39 mutation may partially interfere with CP T41 phosphorylation functions in vivo and reduce the accumulation of virus particles.
Because RNA packaging can control the size and shape of virions21, we carried out experiments to examine whether the small aberrant T41 particles in the preparations contain viral RNAs, by electrophoresing the same amounts of partially purified particles under non-denaturing conditions, followed by staining of the gels with ethidium bromide (EtBr)38,40. Only particles formed by BBSVwt and BBSVR39A contained viral gRNAs, as indicated by positive EtBr staining (Fig. 6c, lanes 1, 3 and 6), whereas lanes containing the small virus-like particles formed by the T41A and T41E mutants did not exhibit detectable EtBr staining (Fig. 6c, lanes 4 and 5). These results strongly suggest that the enriched T41A and T41E mutant particles do not contain viral RNA.
To further evaluate whether viral RNAs are stable after extraction, an RNase-sensitivity assay was carried out by incubating virus-inoculated leaf sap for 30 and 60 min at 37 °C (Fig. 7a). As expected of stable virions, the results showed that, BBSVwt in incubated leaf sap preparations contained the same amounts of viral RNA as extractions made immediately after grinding (Fig. 7a, lanes 1–3). However, after incubation, BBSVT41A (Fig. 7a, lanes 4–6) and BBSVT41E (Fig. 7a, lanes 7–9) gRNAs were degraded by endogenous host RNases present in plant sap. This provides additional evidence that the aberrant virus particles are deficient in morphogenesis steps possibly involved in RNA packaging.

RNA stability in N. benthamiana extracts and RNA-binding affinities of wt and T41 mutants.
(a) RNase protection assay in extracts from N. benthamiana infected with BBSVwt and T41 mutants. Inoculated leaves were ground and incubated in PIPE buffer at 37 °C for 30 (lanes 2, 5 and 8) or 60 mins (lanes 3, 6 and 9) to permit endogenous RNase degradation of unprotected RNA. The total RNAs were subsequently extracted and analyzed by Northern blot with a BBSV-specific probe. Bands corresponding to gRNAs and sgRNAs are indicated on the left. Total RNA was extracted as an untreated control immediately after leaves were ground (lanes 1, 4 and 7). (b) RNA binding abilities of rCPwt (lane 1), rCPT41A (lane 2) and rCPT41E (lane 3) determined by North-western blot assays with an anti-digoxigenin antibody (α DIG, top panel). BSA served as a negative control (lane 4). The bottom panel shows Coomassie blue stained protein loading controls.
Therefore, we carried out a North-western blot assay16 to test whether CP phosphorylation at T41 might affect RNA binding affinity. Compared with the BSA negative control (Fig. 7b, top panel, lane 4), the rCPwt displayed a strong binding affinity for viral RNA (Fig. 7b, top panel, lane l). However, the rCPwt, rCPT41A and rCPT41E (Fig. 7b, top panel, lanes 1–3) each had similar RNA-binding affinities. These results thus suggest that phosphorylation of the T41 residue does not directly affect CP RNA binding activity and that the failure to encapsidate viral RNA occurs at other steps in morphogenesis of the aberrant particles.
Discussion
Protein phosphorylation, which regulates signal transduction, biological activity, molecular interaction and subcellular localization34, is one of the most common posttranslational modifications. Several proteins involved in viral movement are known to be phosphorylated by host kinases, such as the movement proteins (MPs) of the Tobamoviruses, Tomato mosaic virus (ToMV) and Tobacco mosaic virus (TMV)41,42,43,44 and the MPs of Alfalfa mosaic virus (AMV) and Cucumber mosaic virus (CMV), which belong to the family Bromoviridae45,46. More complex movement proteins are also phosphorylated including the triple gene block 1 (TGB1) protein of Potato virus X (PVX), in the family Alphaflexiviridae47 and TGB1s of Potato mop top virus (PMTV) and Poa semilatent virus (PSLV) in the family Virgaviridae48,49. Phosphorylation of these MPs by host kinases has been shown to important for virus cell-to-cell movement through regulation of MP stability42, cellular localization42, RNA binding affinity48 and interactions with other MP partners44,49. These findings suggest that protein phosphorylation has important but poorly understood roles in a number of viral MP processes50,51.
In addition to the nonstructural MPs, viral coat proteins (CPs) involved in virion or RNP assembly can also affect virus movement through phosphorylation with host serine (S)/threonine (T) kinases. As examples, phosphorylation of CPs of helical PVA and PVX regulates disassembly of RNP or virion to permit replication and translation of viral genomic RNA26,27,28,33. However, little is known about the effects of phosphorylation on virion assembly or stability of spherical plant viruses. Our results shown here provide new insight into further understanding of the connection between phosphorylation of the BBSV CP and icosahedral virion assembly.
We found that CP of BBSV is phosphorylated in N. benthamiana at four residues, of which only T41 plays a dominant role in viral long-distance movement (Fig. 2). According to the phosphorylation predictions from the KinasePhos online server and GPS 2.1 program, T41 is phosphorylated by PKA. Furthermore, in vitro PKA phosphorylation assays indicated that mutations of T41 greatly reduced the CP phosphorylation levels (Fig. 3e). It is plausible that the residual signal is due to phosphorylation of other residues (Fig. 3e, lane 4) and we have found by LC-MS/MS that S15 is also located within a PKA consensus site (Fig. 1b). To further investigate CP phosphorylation, S15A, S12/S15/T18/T41A as well as two negative control mutants, S12A and T18A were constructed. The in vitro PKA phosphorylation results with the purified mutant rCPs showed that BBSV CP phosphorylation was reduced significantly in the S15A substitution, compared with that of the rCPwt and the S12A and T18A substitutions (Supplementary Fig. S2, lanes 3, 4 and 6) and incorporation nearly disappeared in the S12/S15/T18/T41A multiple substitution (Supplementary Fig. S2, lanes 5 and 9), Thus, we conclude that S15 residue is also sensitive to PKA. However, the S15 phosphorylation deficient mutant virus has the same local and systemic movement phenotype as BBSVwt suggesting that S15 phosphorylation is not required for long distance movement (Fig. 2, lanes 2 and 4).
Further inoculation analyses suggested that T41 mutations has only minor effects on the replication and translation of viral RNA at 20 hpi of protoplasts, but these mutants had reduced levels of CP and RNA accumulation at 60 hpi (Fig. 5). The T41 mutants also accumulated to lower levels than BBSVwt in inoculated leaves (Fig. 4a–d) and failed to systematically invade upper uninoculated N. benthamiana leaves. (Fig. 4e–h).
Examination of particles in leaves inoculated with the BBSVT41A and BBSVT41E mutant viruses revealed that unlike the virions produced by BBSVwt, the T41 mutant viruses elicited small irregular spheres that did not contain viral RNA (Fig. 6a,c). Previous studies have shown that RNA folding is a factor that can control particle size and shape21,52. Thus, the failure to encapsidate gRNA observed in our experiments (Fig. 6c, lanes 4 and 5) could lead to formation of aberrant virus particles. Moreover, RNase-sensitivity assays suggested that gRNAs of the T41A and T41E mutant viruses are susceptible to degradation by endogenous RNases in N. benthamiana sap (Fig. 7a). These results indicate that CP T41 mutants are unable to package viral RNAs into intact viral particles or protect against RNA degradation.
We initially hypothesized that the failure to encapsidate RNA might be a consequence of RNA binding defects of the CP mutants. However, the negatively charged mutant (rCPT41E) (Fig. 7b, lane 3) displayed similar RNA-binding affinity as that of the non-phosphorylated rCPwt and rCPT41A purified from E. coli (Fig. 7b, lanes 1 and 2). This finding is not consistent with previous research on the helical PVA and BaMV virions or RNP in which CP phosphorylation reduces RNA binding activity27,28,33,53. However, BBSV virions differ from helical viruses in having an icosahedral T = 3 structure in which the major RNA binding domain is located at the basic N-terminal ARM (arginine-rich motif) region16. Given that T41 is located 28 amino acids downstream of the major ARM (4KRNKGGKKSR13) domain16, it is not expected to involved directly in RNA binding. Thus, the most logical hypotheses is that phosphorylation of T41 may be indirectly affect RNA binding during packaging, or that phosphorylation may influence dynamic secondary and tertiary precapsid alterations occurring during virus particle morphogenesis.
Several studies have provided evidence that the N-termini of the CPs of some viruses, such as Sesbania mosaic virus (SeMV), Tomato bush stunt virus (TBSV), CNV and Hibiscus chlorotic ringspot virus (HCRSV) affect the size of the virus particles54,55,56,57. For example, the N-terminal ARM and β-annulus are crucial to T = 3 virion assembly of CNV and deletion of these important regions results in the formation of T = 1 virus-like particles58. Cryo-transmission electron microscopy studies of CNV T = 1 and T = 3 virus particles reveal that T = 3 intact virion is composed of two types of CP dimers which are C-C and A-B subunit dimers. However, T = 1 virus-like particle only contains a single subunit dimer that is very similar to the A-B subunit dimers found in T = 3 particles. Further analyses demonstrate that the S domain of the C-C dimer have a flat conformation with an ordered N-terminal ARM and β-annulus at the internal surface of particle for RNA packaging, whereas the S domain of A-B dimer is more angled and provides limited space at the N-terminus, which tends to be disordered and limits RNA binding. Although RNA binding plays an important role in regulating the A-B and C-C dimers conformations of CNV, TBSV and hepatitis E virus (HEV) particles52,56,58, interactions with host factors such as kinases may be involved in stable T = 3 particle assembly. Support for this hypothesis comes from several recent findings with hepatitis B virus (HBV)59,60,61. PKA phosphorylation at the N-terminus of HBV core protein accelerates capsid assembly and is thought to elicit changes in core protein conformation during assembly59. The HBV core protein is also phosphorylated by protein kinase C (PKC), which affects core protein subunit affinities, capsid assembly and stability60. Thus, phosphorylation is predicted to alter and stabilize the HBV core protein structure during secondary and tertiary interactions that enhance stability of nascent virions59,60,61.
With BBSV, we have carried out an analysis of the crystal structure of an unphosphorylated T = 1 virus-like particle assembled from rCP purified from E. coli (unpublished data). Consistent with the CP of another necrovirus TNV15, the BBSV CP contains an N-terminal R domain, an H domain and a C-terminal S domain. Additionally, like most of T = 1 virus particles in Tombusviridae52,56,58, we have determined that the N-terminal regions of CP subunits in the BBSV T = 1 particle are flexible and disordered, so can not be visualized. However, the T41 residue is visible and is located at a loop region in the H domain that connects the S domain and the β-annulus. We speculate that in the host environment, phosphorylation at T41 might provide a molecular switch that participates in conformation changes of the N-terminus from a disordered state (A-B dimer) to an ordered state (C-C dimer) and that these changes facilitate RNA binding by the N-termini of the C-C dimers and permit final T = 3 virion morphogenesis via integration with A-B dimers. It is easy to image that if all of the dimers have the C-C conformation, the redundant ordered N-termini would induce collision in the nascent particles and might interfere with RNA-binding and assembly, as well as steps involved T = 3 virion morphogenesis. This hypothesis is supported by the results that both the T41A and T41E mutants formed incompletely-assembled particles that resemble T = 1 virions in inoculated leaves and these enriched particles did not contain gRNA. Hence, it is tempting to speculate that dynamic phosphorylation and de-phosphorylation events are required for morphogenesis of T = 3 virions.
We have developed an in vitro particle assembly assay in preliminary attempts to verify our hypotheses40. Unfortunately, in the initial experiments, both PKA phosphorylated rCPwt (Supplementary Fig. S3, C) and non-phosphorylated rCPwt (Supplementary Fig. S3, B) assembled into aberrant virus-like particles with a diameter of 16.5–16.7 nm in the presence of BBSV gRNA. Therefore, in subsequent experiments, we are planning to test viral assembly intermediates that occur during different in vitro environments to provide more rigorous analyses of our models for host factor interactions and specific chemical environments that may be required for intracellular virion assembly. Taken together, our current hypotheses are that the formation and stability of intact T = 3 particles of BBSV are likely dependent on the complex interactions at the N-terminal that may involve host factors and phosphorylation/de-phosphorylation cycles at T41 to provide molecular switches for virion assembly.
In summary, our study is unique in that it provides the first clues suggesting that phosphorylation of the BBSV CP by a host PKA-like kinase has a critical role in viral morphogenesis, which has not been previously reported in the family Tombusviridae. Our data support the notion that the N terminus has important roles in particle assembly and stability. We also hypothesize that the aberrant particles isolated from plants infected with T41 mutants represent T = 1 assembly structures without viral RNA that were blocked at an intermediate stage of morphogenesis. These incompletely assembled and unstable particles do not support long-distance movement of BBSV and this provides another avenue for future research.
Methods
Immunoprecipitation (IP) and detection of CP phosphorylation
Two grams of inoculated N. benthamiana leaves infected with BBSVwt were ground in liquid nitrogen and the resulted powder was dissolved in two volume (w/v) of IP buffer [50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, EDTA-free protease inhibitor cocktail (Sigma), phosphatase inhibitor (Roche), 10% glycerol]. The homogenate was centrifuged at 4 °C for 30 min at 10,000 g and the supernatant was subjected to two additional centrifugation steps. The final supernatant was incubated with CP-specific antibody (1:2000) at 4 °C overnight and further enriched by addition of protein G agarose (Millipore) at 4 °C for 4 hours. The immunoprecipiated CP was used for liquid chromatography-tandem mass spectrometry (LC-MS/MS) and immunoblot analyses.
Identification of CP phosphorylation by LC-MS/MS and immunoblot analyses
For LC-MS/MS identification, immunoprecipitated CP extracted from BBSV inoculated leaf tissues was separated by 12.5% SDS-PAGE and stained with Coomassie brilliant blue R250 (Sigma). After in-gel trypsin digestion and enrichment, the phospho-peptides were separated by nanoscale C18 reverse phase liquid chromatography (Waters) and then electro-sprayed into a Q-Exactive mass spectrometer (Thermo) at the Mass Spectrometry Facility of China Agricultural University. For immunoblot analyses, immunoprecipitated CP was separated by SDS-PAGE and transferred to nitrocellulose membranes (Amersham). A phospho-(Ser/Thr) PKA substrate antibody (Cell Signaling Technology) was used to detect in vivo phosphorylation of CP.
Constructions of plasmids
Several infectious clones used in this study were derived from the infectious cDNA clone of BBSV Ningxia isolate (pUBF52)11. To generate phosphorylation mutants, Quick-change site-directed mutagenesis was used with self-complementary primers (Supplementary Table S1). For expression of CP and its mutants for in vitro phosphorylation assays, wt and mutant fragments were amplified with appropriate primers listed in Supplementary Table S1 and cloned into the pET-30a (+) vector (Novagen). To prepare specific substrates for selected kinases, the oligos corresponding to peptides “RRADDSDDDDD” for CKII, “LRRASLG” for PKA and “KKALRRQETVDAL” for CaMKII were inserted into the pGEX-KG vector62 with an N-terminal glutathione S-transferase (GST) tag using reverse PCR primers (Supplementary Table S1).
Virus inoculation and molecular analysis of progeny viruses
Mechanical inoculation of various mutants onto 2-week-old N. benthamiana plants was performed according to a previous study11, except that all the in vitro transcripts were adjusted to equal amounts by UV spectrophotometry to ensure uniformity of the experiments. At 12 and 24 dpi, total RNA and protein was extracted from infected leaves and prepared for further analyses.
For protoplast transfections, mesophyll protoplasts were prepared from the second and third true leaves of 3 to 4-week-old N. benthamiana63. Approximately 106 isolated protoplasts were transfected with 20 μg of purified transcripts according to a PEG-calcium-mediated transfection method39. After culturing for 20 or 60 hours, total RNA was extracted from the transfected protoplasts with Trizol reagent (Invitrogen) and prepared for further analyses.
Northern blots were conducted according to the manufacturer instructions (DIG high prime DNA labeling and detection kit I, Roche) and DIG-labeled probe was prepared from a DNA fragment corresponding to the 3¢ UTR of the BBSV genome12. Western blots and (enzyme-linked immunosorbent assays) ELISA were performed using CP-specific antibodies as described previously64.
Expression and purification of recombinant protein
E. coli strain BL21 (DE3) pLysS cells (Novagen) were transformed with pET30a-CPwt or phosphorylation mutant constructs by heat shock and cultured at 37 °C until the OD600 reached 0.4. Then, 0.2 mM IPTG (Sigma) was used to induce protein over-expression at 18 °C for another 18 hours. Bacteria cells were then harvested, resuspended in T buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 1 mM PMSF) containing 0.1% Triton X-100 and disrupted by ultrasonication. After centrifugation for 20 min at 20,000 g, the supernatant was collected to purify the N-terminal hexahistidine tagged CP by Ni-NTA agarose affinity (Bio-Rad). After washing with increasing step-wise concentrations of imidazole, proteins eluting in T buffer containing 400 mM imidazole were concentrated with 30 kDa Amicon-Ultra-15 filters (Millipore).
Three pGEX-KG60 based constructs of kinase specific substrates (subCKII, subPKA and subCaMKII) were transformed into E. coli strain BL21 (DE3) pLysS cells. After culturing and induction, proteins were purified from the cells by affinity chromatography on glutathione affinity sepharose columns (GE healthcare). GST-tagged proteins were eluted by T buffer containing 2 mM DTT and 60 mM L-Glutathione and concentrated with Amicon-Ultra-15 filters (30 kDa). Purified protein concentrations were determined by measuring the OD280 with a NanoDrop ND1000 spectrophotometer and proteins were assessed by SDS-PAGE.
In vitro phosphorylation assays
Five μg of rCP or kinase specific substrates and 0.5 μg of kinase [purified NbCKII or commercial mammal kinases PKA or CaMKII (New England Biolabs)], were incubated in 10 μl reactions containing 1X kinase reaction buffer (New England Biolabs), 3 μCi γ-32P-ATP (Perkin Elmer) and 200 μM unlabeled ATP. To inhibit PKA phosphorylation, 1 μM of PKA specific inhibitor H-89 [5-Isoquinolinesulfonamide, N-(2-((3-(4-bromophenyl)-2-propenyl)amino)ethyl)] was added. After incubation at 30 °C for 30 min, the reactions were quenched by addition of 5X SDS sample buffer (250 mM Tris-HCl, pH 6.8, 10% SDS, 50% Glycerol, 0.5% Bromophenol blue, 5% β-mercaptoethanol) and boiled for 5 min. Then samples were separated by 12.5% SDS-PAGE followed by autoradiography.
Virus particle purification
Three procedures were used to obtain BBSVwt and mutant viral particles for Transmission electron microscopy (TEM), Western blots and agarose gel electrophoresis analyses.
To rapidly obtain viral particles, virus particles in clarified leaf sap were extracted from one gram of N. benthamiana leaves inoculated with BBSVwt or mutant viruses at 14 dpi by grinding in 200 μl of 20 mM PBS (pH 7.0) followed by centrifugation at 4 °C for 30 min at 6,000 g. The supernatants were collected and used for various analyses described in the Results.
A previously described partial purification procedure was used to concentrate virus particles38. Briefly, 0.5 gram of virus-inoculated leaves were collected, ground in liquid nitrogen, resuspended at 4 °C in 2 ml of 0.1 M sodium acetate buffer (pH 5.0) containing 5 mM β-mercaptoethanol and the mixture was centrifuged at 4 °C for 30 min at 13,000 g. Then the supernatant was adjusted to 8% PEG6000 and gently shaken for 30 min at 4 °C. The homogenate was centrifuged at 4 °C for 30 min at 13,000 g and the pellet containing viral particles were resuspended in 50 μl ddH2O. For subsequent analyses to visualize virus particles, 5 μl of partial purified particles were subjected to Western blots with a CP-specific antibody. To detect viral gRNA, 2 μg particles were separated by 1% agarose gel in electrophoresis buffer (0.1 M sodium acetate, pH 6.0, 1 mM EDTA) and stained with EtBr.
To obtain more entensively purified viral particles, a conventional and refined purification procedure was performed as described earlier16. Ten grams of virus-inoculated leaves were collected and ground in a blender with two volumes (w/v) of 0.2 M sodium phosphate buffer (pH 7.0) containing 0.2% β-mercaptoethanol. The mixture was then stirred continuously on ice for one hour and centrifuged for 30 min at 10,000 g to remove the insoluble material. The supernatant was collected, mixed with 15% chloroform and centrifuged at 4 °C for 30 min at 13,000 g. Then the supernatant was collected, mixed with 6% PEG6000 and 3% NaCl, stirred for 2 hours and stored overnight at 4 °C. For virus precipitation, the homogenate was centrifuged for 30 min at 13,000 g and the pellet was thoroughly resuspended in 0.2 M sodium phosphate buffer (pH 7.0) containing 0.1% TritonX-100. The clarified supernatant was centrifuged at 4 °C for 90 min at 32,000 g and the final virion pellet was resuspended in 100 μl ddH2O.
Transmission electron microscopy (TEM)
TEM was carried out according to methods described previously64. Briefly, 200 mesh carbon-coated nickel grids were sequentially incubated with BBSV CP-specific antibody and purified particles, followed by staining of the grids with 1% uranylacetate and visualization with a transmission electron microscope (JEM-1230, JEOL Co. Ltd, Japan) operated at 80 kV.
RNA binding assay (North-western blot assay)
Five μg of purified rCPs or BSA, which served as negative control, were separated by 12.5% SDS-PAGE and transferred to nitrocellulose membrane. The proteins were renatured by incubating the membrane overnight at 4 °C in 20 ml of renaturation buffer (50 mM Tris-HCl, pH 7.5, 0.1%TritonX-100, 10% glycerol, 0.1 mM ZnCl2 and 250 mM KCl). Then, a digoxigenin (DIG)-labeled North-western blot procedure was performed as previously described16.
RNase-sensitivity assays
The stability of virus particles to endogenous RNases in leaf sap was determined by an RNase-sensitivity assay as previously reported16,65 with minor modifications. Briefly, 0.2 gram of virus-infected leaves were ground in liquid nitrogen and incubated in 200 μl PIPES buffer (50 mM PIPES, pH 6.5, 0.1% Tween20) at 37 °C for 30 or 60 min. Then, total RNA was extracted using Trizol reagent. Untreated samples, whose total RNA was extracted immediately served as controls. The amount of remaining viral RNA was analyzed by Northern blot as described above.
Additional Information
How to cite this article: Zhao, X. et al. Phosphorylation of Beet black scorch virus coat protein by PKA is required for assembly and stability of virus particles. Sci. Rep. 5, 11585; doi: 10.1038/srep11585 (2015).
References
Cui, X. An icosahedral virus found in sugar beet. J. Shihezi Agric. College 10, 73–78 (1988).
Merhvar, M. & Bragard, C. Beet black scorch virus in Iran is more diverse than anywhere. Phytopathol. 99, S84–S85 (2009).
Weiland, J. J. et al. Characterization of a US Isolate of Beet black scorch virus. Phytopathol. 97, 1245–1254 (2007).
Weiland, J. J., Larson, R. L., Freeman, T. P. & Edwards, M. C. First report of Beet black scorch virus in the United States. Plant Dis. 90, 828–828 (2006).
Koenig, R. & Valizadeh, J. Molecular and serological characterization of an Iranian isolate of Beet black scorch virus. Arch. Virol. 153, 1397–1400 (2008).
González-Vázquez, M., Ayala, J., García-Arenal, F. & Fraile, A. Occurrence of Beet black scorch virus infecting sugar beet in Europe. Plant Dis. 93, 21–24 (2009).
Cai, Z. et al. Identification of pathogenic virus of beet black scorch disease and detection by synthesized cDNA probes. J. Beijing Agric. Univ. 19, 112 (1993).
Jiang, J. et al. Transmission of Beet black scorch virus by Olpidium brassicae. J. Jiangxi Agric. Univ. 21, 525–528 (1999).
Xu, J. et al. Two distinct sites are essential for virulent infection and support of variant satellite RNA replication in spontaneous Beet black scorch virus variants. J. Gen. Virol. 93, 2718–2728 (2012).
Guo, L. H. et al. Analysis of nucleotide sequences and multimeric forms of a novel satellite RNA associated with Beet black scorch virus. J. Virol. 79, 3664–3674 (2005).
Yuan, X. et al. Analysis of the subgenomic RNAs and the small open reading frames of Beet black scorch virus. J. Gen. Virol. 87, 3077–3086 (2006).
Cao, Y. et al. Effect of Beet black scorch virus coat protein on viral pathogenicity. Prog. Biochem. Biophys. 33, 127–134 (2006).
Cao, Y. et al. The complete nucleotide sequence of Beet black scorch virus (BBSV), a new member of the genus Necrovirus. Arch. Virol. 147, 2431–2435 (2002).
Wada, Y. et al. The structure of Melon necrotic spot virus determined at 2.8 Å resolution. Acta Crystallogr. Sect F Struct. Biol. Cryst. Commun. 64, 8–13 (2008).
Oda, Y. et al. Crystal structure of Tobacco necrosis virus at 2.25 Å resolution. J Mol. Biol. 300, 153–169 (2000).
Zhang, X. et al. N-terminal basic amino acid residues of Beet black scorch virus capsid protein play a critical role in virion assembly and systemic movement. Virology 10, 200 (2013).
Reade, R., Kakani, K. & Rochon, D. A highly basic KGKKGK sequence in the RNA-binding domain of the Cucumber necrosis virus coat protein is associated with encapsidation of full-length CNV RNA during infection. Virology 403, 181–188 (2010).
Park, S. H., Sit, T. L., Kim, K. H. & Lommel, S. A. The Red clover necrotic mosaic virus capsid protein N-terminal amino acids possess specific RNA binding activity and are required for stable virion assembly. Virus Res. 176, 107–118 (2013).
Rao, A. L. N., Chaturvedi, S. & Garmann, R. F. Integration of replication and assembly of infectious virions in plant RNA viruses. Curr. Opin. Virol. 9, 61–66 (2014).
Rao, A. L. N. Genome packaging by spherical plant RNA viruses. Ann. Rev. Phytopathol. 44, 61–87 (2006).
Schneemann, A. The structural and functional role of RNA in icosahedral virus assembly. Ann. Rev. Microbiol. 60, 51–67 (2006).
Zhang, Y. et al. Nuclear localization of Beet black scorch virus capsid protein and its interaction with importin α. Virus Res. 155, 307–315 (2011).
Hipper, C., Brault, V., Ziegler-Graff, V. & Revers, F. Viral and cellular factors involved in phloem transport of plant viruses. Front. Plant Sci. 4, No. 154 (2013).
Chapdelaine, Y., Kirk, D., Karsies, A., Hohn, T. & Leclerc, D. Mutation of capsid protein phosphorylation sites abolishes Cauliflower mosaic virus infectivity. J. Virol. 76, 11748–11752 (2002).
Champagne, J., Laliberté-Gagné, M.-E. & Leclerc, D. Phosphorylation of the termini of Cauliflower mosaic virus precapsid protein is important for productive infection Mol. Plant-Microbe Interact. 20, 648–658 (2007).
Atabekov, J. G. et al. Translational activation of encapsidated Potato virus X RNA by coat protein phosphorylation. Virology 286, 466–474 (2001).
Ivanov, K. I., Puustinen, P., Merits, A., Saarma, M. & Mäkinen, K. Phosphorylation down-regulates the RNA binding function of the coat protein of Potato virus A. J. Mol. Biol. 276, 13530–13540 (2001).
Ivanov, K. I. et al. Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and its relevance for virus infection. Plant Cell 15, 2124–2139 (2003).
Bhat, A. S. & Savithri, H. S. Investigations on the RNA binding and phosphorylation of Groundnut bud necrosis virus nucleocapsid protein. Arch. Virol. 156, 2163–2172 (2011).
Akamatsu, N. et al. Phosphorylation and interaction of the movement and coat proteins of Brome mosaic virus in infected barley protoplasts. Arch. Virol. 152, 2087–2093 (2007).
Fernández-Fernández, M. R. et al. The capsid protein of a plant single-stranded RNA virus is modified by O-linked N-acetylglucosamine. J. Biol. Chem. 277, 135–140 (2002).
Pérez Jde, J. et al. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances viral infection. Virol. 442, 122–131 (2013).
Hung, C. J. et al. Phosphorylation of coat protein by protein kinase CK2 regulates cell-to-cell movement of Bamboo mosaic virus through modulating RNA binding. Mol. Plant-Microbe Interact. 27, 1211–1225 (2014).
Ubersax, J. A. & Ferrell, J. E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Bio. 8, 530–541 (2007).
Xue, Y. et al. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 24, 255–260 (2011).
Huang, H. D., Lee, T. Y., Tzeng, S. W. & Horng, J. T. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 33, W226–229 (2005).
Reuveni, H. et al. Toward a PKB inhibitor: modification of a selective PKA inhibitor by rational design. Biochem. 41, 10304–10314 (2002).
Robbins, M. A., Reade, R. D. & Rochon, D. M. A Cucumber necrosis virus variant deficient in fungal transmissibility contains an altered coat protein shell domain. Virology 234, 138–146 (1997).
Wang, X. et al. The R-rich motif of Beet black scorch virus P7a movement protein is important for the nuclear localization, nucleolar targeting and viral infectivity. Virus Res. 167, 207–218 (2012).
Cadena-Nava, R. D. et al. Self-assembly of viral capsid protein and RNA molecules of different sizes: requirement for a specific high protein/RNA mass ratio. J. Virol. 86, 3318–3326 (2012).
Matsushita, Y., Ohshima, M., Yoshioka, K., Nishiguchi, M. & Nyunoya, H. The catalytic subunit of protein kinase CK2 phosphorylates in vitro the movement protein of Tomato mosaic virus. J. Gen. Virol. 84, 497–505 (2003).
Kawakami, S. et al. Phosphorylation and/or presence of serine 37 in the movement protein of Tomato mosaic tobamovirus is essential for intracellular localization and stability in vivo. J. Virol. 73, 6831 (1999).
Karger, E. M. et al. Dysfunctionality of a Tobacco mosaic virus movement protein mutant mimicking threonine 104 phosphorylation. J. Gen. Virol. 84, 727–732 (2003).
Waigmann, E., Chen, M.-H., Bachmaier, R., Ghoshroy, S. & Citovsky, V. Regulation of plasmodesmal transport by phosphorylation of Tobacco mosaic virus. EMBO J. 19, 4875–4884 (2000).
Matsushita, Y., Yoshioka, K., Shigyo, T., Takahashi, H. & Nyunoya, H. Phosphorylation of the movement protein of Cucumber mosaic virus in transgenic tobacco plants. Virus Genes 24, 231–234 (2002).
Kim, B. S., Halk, E. L., Merlo, D. J., Nelson, S. E. & Loesch-Fries, L. S. Phosphorylation of Alfalfa mosaic virus movement protein in vivo. Arch. Virol. 159, 1787–1791 (2014).
Módena, N. A., Zelada, A. M., Conte, F. & Mentaberry, A. Phosphorylation of the TGBp1 movement protein of Potato virus X by a Nicotiana tabacum CK2-like activity. Virus Res. 137, 16–23 (2008).
Makarov, V. V., Iconnikova, A. Y., Guseinov, M. A., Vishnichenko, V. K. & Kalinina, N. O. In vitro phosphorylation of the N-terminal half of hordeivirus movement protein. Biochem. (Moscow) 77, 1072–1081 (2012).
Samuilova, O., Santala, J. & Valkonen, J. P. Tyrosine phosphorylation of the triple gene block protein 3 regulates cell-to-cell movement and protein interactions of Potato mop top virus. J. Virol. 87, 4313–4321 (2013).
Lucas, W. J. Plant viral movement proteins: Agents for cell-to-cell trafficking of viral genomes. Virology 344, 169–184 (2006).
Schoelz, J. E., Harries, P. A. & Nelson, R. S. Intracellular transport of plant viruses: finding the door out of the cell. Mol. Plant. 4, 813–831 (2011).
Xing, L. et al. Structure of hepatitis E virion-sized particle reveals an RNA-dependent viral assembly pathway. J. Biol. Chem. 285, 33175–33183 (2010).
Vijayapalani, P. et al. Phosphorylation of Bamboo mosaic virus satellite RNA (satBaMV)-encoded protein P20 downregulates the formation of satBaMV-P20 ribonucleoprotein complex. Nucl. Acid. Res. 40, 638–6495 (2012).
Hui, E. & Rochon, D. Evaluation of the roles of specific regions of the Cucumber necrosis virus coat protein arm in particle accumulation and fungus transmission. J. Virol. 80, 5968–5975 (2006).
Lokesh, G. L., Gowri, T. D., Satheshkumar, P. S., Murthy, M. R. & Savithri, H. S. A molecular switch in the capsid protein controls the particle polymorphism in an icosahedral virus. Virology 292, 211–223 (2002).
Hsu, C. et al. Characterization of polymorphism displayed by the coat protein mutants of Tomato bushy stunt virus. Virology 349, 222–229 (2006).
Niu, S. et al. Hibiscus chlorotic ringspot virus coat protein is essential for cell-to-cell and long-distance movement but not for viral RNA replication. PLoS ONE 9, e113347 (2014).
Katpally, U. et al. Structures of T = 1 and T = 3 particles of Cucumber Necrosis Virus: evidence of internal scaffolding. J. Mol. Biol. 365, 502–512 (2007).
Kang, H. Y. et al. Phosphorylation of hepatitis B virus Cp at Ser87 facilitates core assembly. Biochem. J. 398, 311–317 (2006).
Kang, H., Yu, J. & Jung, G. Phosphorylation of hepatitis B virus core C-terminally truncated protein (Cp149) by PKC increases capsid assembly and stability. Biochem. J. 416, 47–54 (2008).
Wittkop, L. et al. Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell. Microbio. 12, 962–975 (2010).
Guan, K. & Dixon, J. E. Eukaryotic proteins expressed in Escherichia coli: An improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 192, 262–267 (1991).
Nagy, J. I. & Maliga, P. Callus induction and plant regeneration from mesophyll protoplasts of Nicotiana sylvestris. Z. Pflanzenphysiol. 78, 453–455 (1976).
Zhang, Y. et al. Development of Tobacco necrosis virus A as a vector for efficient and stable expression of FMDV VP1 peptides. Plant Biotechnol. J. 8, 506–523 (2010).
Kaplan, I. B. et al. Point mutations in the Potato leafroll virus major capsid protein alter virion stability and aphid transmission. J. Gen. Virol. 88, 1821–1830 (2007).
Acknowledgements
We thank Dr. Andrew O. Jackson (University of California at Berkeley) for constructive criticism and helpful editorial suggestions, Dr. Yau-Heiu Hsu (National Chung Hsing University) for kindly providing the detail protocol for in vitro phosphorylation assays, Dr. Zhen Li (Mass Spectrometry Facility, China Agricultural University) for technical assistance with LC-MS/MS and Drs. Huiqiang Lou, Dongtao Ren and Qun He (CAU) for helpful suggestions. This work was supported by the National Natural Science Foundation of China (31470253 and 31100115).
Author information
Authors and Affiliations
Contributions
X.Z. and D.L. designed the project and wrote the manuscript, X.Z. performed almost experiments, X.W., K.D., Y.H., X.Z. and Y.C. analyzed the data, Y.Z. conducted the TEM and revised the manuscript, X.W., C.H. and J.Y. contributed through discussions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, X., Wang, X., Dong, K. et al. Phosphorylation of Beet black scorch virus coat protein by PKA is required for assembly and stability of virus particles. Sci Rep 5, 11585 (2015). https://doi.org/10.1038/srep11585
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11585
This article is cited by
-
Plant viral proteins and fibrillarin: the link to complete the infective cycle
Molecular Biology Reports (2021)
-
Distribution and phylogenetic analysis of the 3′UTR and coat protein gene of Iranian Beet black scorch virus
Journal of Plant Diseases and Protection (2019)
-
Proteotype profiling unmasks a viral signalling network essential for poxvirus assembly and transcriptional competence
Nature Microbiology (2018)
-
Hsc70-2 is required for Beet black scorch virus infection through interaction with replication and capsid proteins
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
