
Abstract
The aim of this study was to evaluate the contribution of mitochondrial DNA (mtDNA) mutations in oxidative phosphorylation (OXPHOS) deficiency. The complete mitochondrial genomes of 41 families with OXPHOS deficiency were screened for mutations. Mitochondrial functional analysis was then performed in primary and cybrid cells containing candidate mutations identified during the screening. A novel mitochondrial NADH dehydrogenase 5 (ND5) m.12955A > G mutation was identified in a patient with exercise intolerance and developmental delay. A biochemical analysis revealed deficiencies in the activity of complex I (NADH:quinone oxidoreductase) and IV (cytochrome c oxidase) of this patient. Defects in complexes I and IV were confirmed in transmitochondrial cybrid cells containing the m.12955A > G mutation, suggesting that this mutation impairs complex I assembly, resulting in reduced stability of complex IV. Further functional investigations revealed that mitochondria with the m.12955A > G mutation exhibited lower OXPHOS coupling respiration and adenosine triphosphate (ATP) generation. In addition, the cytotoxic effects, determined as reactive oxygen species (ROS) and lactate levels in the present study, increased in the cells carrying a higher m.12955A > G mutant load. In conclusion, we identified m.12955A > G as a mitochondrial disease-related mutation. Therefore, screening of m.12955A > G is advised for the diagnosis of patients with mitochondrial disease.
Similar content being viewed by others


Introduction
Mitochondria play a central role in a variety of cellular processes, including oxidative phosphorylation (OXPHOS), cell signaling, calcium buffering and apoptosis1. There are approximately 1000 proteins in the mitochondria, only 13 of which are encoded by mitochondrial DNA (mtDNA). Most mitochondrial diseases are caused by nuclear-encoded DNA mutations and follow Mendel’s laws of inheritance2. However, many fatal pathological conditions such as Leigh syndrome, leukoencephalopathy and some degenerative diseases such as cancer, Leber hereditary optic neuropathy (LHON) and mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) are associated with mtDNA mutations2.
Mitochondrial respiratory complex I (NADH:quinone oxidoreductase), which is the first and largest enzyme in OXPHOS, is the major electron entry point to the mitochondrial respiratory chain. A long-term survey of mitochondrial respiratory chain disorders in Australian children showed that OXPHOS disorders were frequently observed in newborns, with an estimated incidence of 1 in 5,000 and complex I defects accounted for approximately 25–35%3,4 of these cases. Complex I is composed of 38 nuclear subunits and seven mtDNA encoded subunits, which are assembled in the inner mitochondrial membrane to form an L-shaped structure. In mammalian cells, complex I consists of 14 core subunits for the assembly of the complex I scaffold motif and 31 additional supernumerary subunits of unknown function5,6. In vertebrates, all seven mtDNA encoded hydrophobic subunits [mitochondrially encoded NADH dehydrogenase 1–6 and 4L (ND1-ND6 and ND4L)] are core subunits of complex I and form the major complex I membrane arm. Mutations in NADH dehydrogenase (ND) subunits can disrupt either complex I assembly or enzyme activity7. Recent studies have shown that approximately 20% of cases of isolated complex I deficiency were caused by mtDNA mutations in mitochondrially encoded NADH dehydrogenase (MTND) genes8. In particular, mutations in ND subunits were associated with LHON, MELAS and Leigh Syndrome9. New pathogenic MTND mutations are being frequently identified, but the functional consequences of these mutations have not been confirmed10,11. Elucidation of the functional effects of pathogenic mtDNA mutations will aid in the genetic diagnosis of patients with complex I deficiency. However, the identification of new pathogenic mtDNA mutations is difficult due to the diversity of mtDNA polymorphisms and the clinical heterogeneity of disease.
In this study, we screened the whole mitochondrial genome in a large cohort of 41 pediatric patients with biochemical manifestations of complex I deficiency. Patients with recurrent mutations such as m.3243A > G, m.8993T > G/C and m.8344A > G were ruled out of the study before the sequence screening. The reported pathogenic mtDNA mutations m.3697G > A and m.14487T > C were identified in two patients with MELAS and Leigh syndrome, respectively12,13. Furthermore, we identified one patient with exercise intolerance and developmental delay harboring a novel pathogenic mutation m.12955A > G [p. Asn(N)207Asp(D)]. To further confirm the pathogenic mechanism of the m.12955A > G mutation, a series of cytoplasmic hybrid (cybrid) cell lines with different loads of the m.12955A > G mutation were constructed by transferring mitochondria from the patient into mtDNA-less ρ0 human osteosarcoma 143B cells. The effects of the m.12955A > G mutation on OXPHOS deficiency was analyzed by assessing mitochondrial respiration, adenosine triphosphate (ATP) content, reactive oxygen species (ROS) levels and lactate acid levels in addition to complex I assembly and activity.
Results
Clinical data
Samples from 41 patients diagnosed with OXPHOS deficiency and their maternal family members were obtained from Peking University First Hospital (China). Informed consent was obtained from all subjects under protocols approved by the Ethical Committee of the Peking University First Hospital. All experimental methods were carried out in accordance with approved guidelines of Peking University First Hospital. The patient (female, aged 9 years at the time of this study) identified with the novel mtDNA 12955A > G mutation, was born in a non-consanguineous Chinese family and presented with clinical manifestations of exercise intolerance and developmental delay. The child was initially investigated for muscle weakness, malnutrition and developmental and psychomotor retardation at the age of 6 months. She was referred to our hospital at the age of 2 years and 3 months owing to her inability to walk. The patient showed progressive deterioration in terms of muscle pain, severe fatigue and dizziness, which were exacerbated by a short period of exercise. Physical examination indicated that the patient had limb hypotonia, muscle relaxation, knee tendon reflex hyperfunction with normal ankle clonus and Babinski sign, as well as reduced growth [weight: 22 kg (normal 24.1–35.3 kg)]; height: 123 cm (normal 125.7–138.7 cm). Magnetic resonance imaging of the brain revealed no pathological alterations. Cerebrospinal fluid (CSF) and blood lactate levels were 1.3 mmol/L (normal range, 1.0–2.8 mmol/L) and 3.9 mmol/L (normal range, 0.5–2 mmol/L), respectively. The lactate/pyruvate ratio in the CSF was 15 but reached 58 in blood (normal range <20), suggesting a respiratory chain dysfunction in this patient. Following treatment with a combination of L-carnitine (1 g/day), calcium folinate (15 mg/day), arginine (1 g/day), vitamin B1 [30 mg twice daily (b.i.d.)], vitamin B2 (5 mg b.i.d.), vitamin C (100 mg/day), vitamin E (50 mg b.i.d.) and coenzyme Q10 (10 mg b.i.d.) at 2 years and 3 months of age, the patient’s symptoms improved significantly and she was able to walk unaided after 3 months of treatment.
Biochemical analysis
The enzyme activity of complex V (ATP synthase) and four mitochondrial respiratory chain complexes (MRCs) were measured in the lymphoblasts of the patient, as previously described14. The activity of each enzyme was normalized against that of citrate synthase, a mitochondrial matrix marker enzyme (Table 1). Based on the criteria of OXPHOS enzyme activity from 100 healthy children, defective complexes I and IV were identified in the patient (Table 1).
Identification of pathogenic mutations
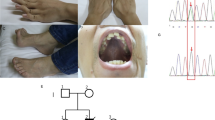
To identify the pathogenic role of these MRCs deficiencies, the whole mitochondrial genome of the patient and her maternal relatives were subjected to Sanger sequencing. The patient was found to be carrying an unreported heteroplasmic mutation, m.12955A > G (p.N207D). The mutant load was quantified by subjecting the blood and urine samples from the patient and a blood sample from her mother to real-time polymerase chain reaction (qPCR). As shown in Fig. 1A, the mutant load was higher in the blood of the patient compared with that in the blood of her mother (50.1% vs. 32%, respectively). In the urinary epithelium of the patient, the mutant load of m.12955A > G was even higher (63.2%). Since blood and urinary epithelium always contain lower mutant loads than do other tissues, we believed that a higher mutant load of m.12955A > G would be found in other tissues such as brain and muscle from the patient15,16. Unfortunately, we were unable to analyze the mutant loads in muscle and fibroblasts from the patient. In addition, the patient’s parents declined further investigations since our diagnosis of mild clinical manifestation was similar to a previous report by Downham et al.17. In addition, no complex IV-related mtDNA mutations were detected, although a decline of complex IV activity was identified in this patient (Table 2). Since the stability and activity of some MRCs are interdependent, it is possible that the complex I deficiency caused by m.12955A > G may decrease the assembly/stability of complex IV18,19. Furthermore, the possibility of a rare polymorphism at 12955 was excluded by screening for the mutation in 161 control subjects.

Family tree and cell models of m.12955A>G.
(A) The proband is indicated by an arrow; mutant loads of lymphocytes (L) and urinary epithelium (U) were determined by real-time PCR. (B) Sequence diagrams of clone #9, #17 and #18 cells, N: Asn, D: Asp. (C) Morphology of cells after culturing for 24 h; 143B wild type (WT): cells without mtDNA mutation.
The features of m.12955A > G were further analyzed to evaluate its pathogenicity. A protein structure model of ND5 revealed that residue 207 is located in the third hydrophilic extramembrane loop (http://www.uniprot.org/uniprot/P03915). An N to D substitution introduces a negative charge at residue 207 in the extramembrane loop, thereby changing the electrostatic forces, which are important for protein-protein interactions20. Together, these findings indicated the pathogenic potential of the m.12955A > G mutation.
Construction of cybrid cell lines containing different m.12955A>G mutant loads
To understand the pathogenic role of m.12955A > G fully, we generated a series of cybrid cell lines containing different loads of the mutation. Briefly, platelets from the patient were fused with mtDNA-less ρ0 human osteosarcoma 143B cells and 20 single cybrid clones were selected by culturing the fusion mixtures in medium without uridine and sodium pyruvate for 15 days. Clone #9, #17 and #18 cells with mutant loads of 65.21%, 21.76% and 98.14%, respectively, were selected by real-time PCR for further analysis. However, owing to the poor sensitivity of Sanger sequencing in detecting of heteroplasmic mutations at mutant loads below 20%, the mutation was not detected in the sequencing of clone #17 (Fig. 1B).
Analysis of the cell morphology of the selected clones revealed that increased loads of the m.12955A > G mutation were associated with decreased cell viability (Fig. 1C). Cells with 20% of the m.12955A > G mutation had a smooth cell membrane and an elongated cell shape, which were comparable with that observed in wild type 143B cells, while cells with 65% of the heteroplasmic m.12955A > G mutation were smaller. Furthermore, cells with 98% of heteroplasmic m.12955A > G cells were aggregated and showed reduced viability (Fig. 1C). This result indicates that an m.12955A > G mutation affects cell viability in a mutant load-dependent manner. Due to the similar morphology between clone #17 cells and wild type 143B cells and the observation that the m.12955A > G mutant load in the mother of the patient was approximately 30% but without the clinical phenotype, clone #17 cells were used as a control for clone #9 and #18 cells.
Blue native PAGE analysis of respiratory complex assembly
Intact complex I was analyzed by blue native polyacrylamide gel electrophoresis (BN-PAGE) to evaluate whether the m.12955A > G mutation impairs the assembly of complex I. Mitochondria isolated from the cells of clone #9, #17 and #18 were separated by BN-PAGE to assay for in-gel complex I activity and immunoblotting analysis of complex I with an antibody against NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 13 (NDUFA13). As shown in Fig. 2A, cells of clone #18 cells had less intact complex I but more subcomplex I just below the position of complex I, compared to that observed in the cells of clone #9 and #17. However, complex I assembly in clone #9 was not affected when compared with clone #17 cells, suggesting that complex I assembly was impaired only by high m.12955A > G mutant loads. Moreover, a considerable decrease in complex IV assembly was found in clone #18 cells compared with clone #17 cells, while assembly of complex IV was normal in clone #9 cells. This indicates that the decrease in complex I assembly in clone #9 cells is not sufficient to affect complex IV assembly. Additionally, the assembly of complex II (succinate dehydrogenase), complex III (ubiquinol-cytochrome c reductase) and complex V were not affected in either clone #9 or #18 cells, compared with clone #17 cells (Fig. 2A). In-gel activity assays confirmed that the nearly homoplasmic mutant cybrid cells of clone #18 cells exhibited a significantly reduced complex I and IV activity relative to that of clone #17 cells (Fig. 2B). Together, our results indicate that a high m.12955A > G mutant load can impair complex I assembly, which may further affect the stability of complex IV.

Respiratory complex assembly and in-gel activity assay.
(A) Whole cells were solubilized with n-dodecyl-β-D-maltoside (DDM) and then subjected to BN-PAGE/immunoblot analysis. Complexes I, II, III, IV and V were immunoblotted with anti-Grim19, SDHA, core2, COX IV and ATP-5A antibodies, respectively; β-VDAC was used as internal control. (B) Mitochondria were solubilized with DDM. In-gel activity assays of complexes I and IV were performed following BN-PAGE analysis; the VDAC protein was used as a loading control. All BN-PAGEs were run under the same conditions (5 mA per gel for 1 h and followed by electrophoresis at 200 V for 2 h). Full-length blots are presented in Supplementary Figs. 1 and 2. Error bars, ±SD *, P < 0.05.
Mitochondrial functional analysis
The endogenous respiration of intact cells was first measured to clarify the role of increased m.12955A > G mutant loads on mitochondrial function. As shown in Fig. 3A, although no significant decrease in base respiration was detected in intact cells, clone #9 and #18 cells showed a trend towards decreased respiratory function. Furthermore, coupled and uncoupled mitochondrial respiration among clone #9, #17 and #18 cells was measured in the presence of oligomycin, which completely blocks the ATPase proton channel. Clone #17 cells exhibited higher coupled OXPHOS respiration and respiratory coupling efficiency compared with clone #9 and #18 cells (Fig. 3A and B). Since only clone #18 cells exhibited a significantly reduced complex I [NADH:nitrotetrazolium blue (NTB) oxidoreductase] activity relative to that of clone #17 cells in in-gel activity assay (Fig. 2B), complex I activity of clone #9 cells was further measured as NADH:ubiquinone oxidoreductase using spectrophotometry-based complex I enzyme activity assay. As shown in supplementary Fig. 3, clone #9 cells exhibited a significantly reduced complex I (NADH:ubiquinone oxidoreductase) activity compared with control #17 cells. These observations suggested that the m.12955A > G mutation in ND5 impaired mitochondrial function by decreasing both the total mitochondrial respiration and the respiratory coupling efficiency.

Respiration assay of cells containing the m.12955A>G mutation.
(A) Mitochondrial respiratory capacities were determined in clone #9, #17 and #18 cells. Oligomycin (2.5 μg/ml) was added for the measurement of uncoupled mitochondrial respiration. OXPHOS coupling respiration was calculated by subtracting the uncoupled component value from the total endogenous respiration value. (B) Respiratory coupling efficiencies were presented as the ratio of basal oxygen consumption to the oxygen consumption with oligomycin. Error bars, ±SD. *, P < 0.05;
To further confirm the effect of m.12955A > G on mitochondrial function, ATP synthesis was determined in boiled cells by luminometry. As shown in Figure 4A, although the total ATP content in cells that were 65% heteroplasmic (clone #9) were not significant different with those in cells that were 21% heteroplasmic (clone #17), clone #9 cells showed a trend towards decreased ATP generation. Furthermore, the ATP generation in cells that were 98% heteroplasmic (clone #18) was much lower, suggesting a positive correlation between mitochondrial dysfunction and m.12955A > G mutant load. After the addition of rotenone, the ATP generation by respiratory complex I decreased with an increasing m.12955A > G mutant load (Fig. 4A). Thus, our findings further confirm that m.12955A > G impairs mitochondrial function by affecting complex I.

Mitochondrial function and cytotoxicity.
(A) Total ATP content was measured in clone #9, #17 and #18 cells; the rotenone-resistant ATP content was determined in cells treated with 200 nM rotenone for 24 h. The rotenone-sensitive (RS) ATP production was calculated by subtracting the rotenone-resistant component from the total ATP content. (B) Extracellular lactate levels were measured in the medium after 48 h in culture. (C) Mitochondrial ROS were determined in clone #9, #17 and #18 cells; complex I-dependent ROS levels were measured in cells exposed to 1 μM rotenone for 1 h. The values for ATP, lactate and ROS were normalized by protein concentration. Error bars, ±SD. *, P < 0.01; **, P < 0.001.
To test the toxicity of the m.12955A > G mutation, extracellular lactate acid and mitochondrial ROS levels were measured in all three cell lines. As shown in Fig. 4B, lactate acid generation increased with the mutant load of m.12955A > G. Furthermore, a significant increase in mitochondrial ROS was detected in clone #18 cells compared with that in clone #17 (Fig. 4C). After treatment with the complex I-related ROS-inducer rotenone for 1 h, both clone #9 and #17 cells showed a dramatic increase (approximately 1 fold for both) in ROS generation, while the increase in clone #18 cells was slight (approximately 20%). Interestingly, the ROS levels induced by rotenone in clone #9 and #17 cells were comparable with those in clone #18 cells with or without rotenone. This indicated that the high ROS levels in clone #18 cells were caused by the impairment of complex I.
To clearly understand the implications of m.12955A > G in mitochondrial dysfunction, the effect of the mutation in mitochondrial function was also determined by comparing clone #9, #17 and #18 with 143B wild type cells. As shown in Supplementary Fig. 4A, we found that both clone #9 and #18 cells presented a significant decrease of complex I-related ATP generation relative to that in 143B wild type cells. Clone #17 cells showed a mild decrease of complex I related ATP generation when compared to 143B wild type cells. In Supplementary Fig. 4B, the toxicity test for the m.12955A > G mutation showed that clone #9, #17 and #18 cells exhibited higher mitochondrial ROS generation than did 143B wild type cells. After the addition of rotenone, the mitochondrial ROS levels were more or less the same in the four cell lines, suggesting that the m.12955A > G mutation increases mitochondrial ROS generation through the impairment of complex I. Taken together, our results confirm that the mutation of m.12955A > G in complex I causes disease.
Discussion
In the present study, we have identified a novel missense m.12955A > G mutation in the mitochondrial ND5 gene from a patient diagnosed with exercise intolerance and developmental delay. To evaluate the casual role of the m.12955A > G mutation in disease pathogenesis, a detailed analysis of the mutation at the clinical, genetic and molecular levels was performed. As shown in the case report, the patient with a high level of the 12955A > G mutation exhibited a metabolic defect that was partially alleviated by the administration of a combination of drugs for the treatment of OXPHOS deficiency. Together with the fact that the m.12955A > G mutant load in the patient was much higher than that in her mother, this observation suggested a possible correlation with disease occurrence. Unfortunately, we were unable to analyze the mutant loads in muscle and fibroblast cells from the patient as the patient’s parents did not allow further investigations. However, the heteroplasmy of mtDNA mutations are usually higher in muscle and fibroblast cells than in the blood and urinary epithelium21. Genetically, m.12955A > G was the only variation in the patient that was not listed in the mtDNA polymorphism databases from MITOMAP (http://www.mitomap.org), mtDB (http://www.genpat.uu.se/mtDB/) and mtSNP (http://mtsnp.tmig.or.jp). Moreover, we found that m.12955A > G was not detected in 161 Chinese control subjects. Thus, we speculate a possible pathogenic role of 12955A > G in human diseases.
At the molecular level, the activity of mitochondrial complexes I and IV was decreased in cybrid cells containing 98% of the m.12955A > G mutation (clone #18), which was consistent with the biochemical phenotype of the patient. As a result, a mutant load of 98% of m.12955A > G resulted in down-regulated mitochondrial biogenesis by decreasing OXPHOS-coupled mitochondrial respiration and OXPHOS-related ATP generation. Thus, tissues such as neurons and muscle, with a high OXPHOS demand, will be primarily affected. On the other hand, the mutant load of 98% m.12955A > G increased cytotoxicity by elevating both mitochondrial ROS and lactate acid levels. Although the intact respiratory complex levels and ROS generation were not affected in the 65% heteroplasmic 12955A > G cells, the decrease in respiratory coupling efficiency was comparable with that of the homoplasmic mutant (98%) cells. Functional studies further confirmed a trend towards lower ATP generation and a significant increase in lactate acid levels in the cells with 65% heteroplasmic of m.12955A > G compared with that of the cells with 21% heteroplasmic of m.12955A > G. All these above results strongly support the hypothesis that the m.12955A > G mutation is causative and that the severity of the disease is largely dependent on the mutant load.
ND5 is one of 14 essential subunits that constitute the backbone of respiratory complex I5. Deficiencies in ND1, ND2, ND4 and ND6 are proposed to disrupt complex I assembly, while defects in ND3 and ND5 have modest effects on complex I assembly but marked effects on enzyme activity9,22,23. Previously, we demonstrated that mitochondrial respiration is tightly controlled by ND5 by using a cell model with a nonsense ND5 mutation24. Here, only a mild decrease in endogenous oxygen consumption was observed in cells containing both 65% and 98% mutant loads of m.12955A > G compared with endogenous oxygen consumption in cells containing 21% mutant loads of m.12955A > G. However, the OXPHOS-coupled respiration was dramatically decreased in these two cybrid cell lines. Determination of the crystal structure of the bacterial complex I indicate that NADH-quinone oxidoreductase subunit 12 (Nqo12), a homolog of human ND5, is composed of a putative proton translocation channel and the residue of 207 is close to the proton-pumping channel and ubiquinol-binding site6. Thus, it can be speculated that the 12955 (p.N207D) mutation is more likely to affect proton pumping and ubiquinol binding directly. This speculation was confirmed in our results that the NADH:ubiquinone oxidoreductase rather than NADH:NTB oxidoreductase of complex I was decreased in clone #9 cells compared with clone #17 cells. In addition, the level of fully assembled complex I is decreased in cells carrying the homoplasmic m.12955A > G mutation, which is consistent with previous reports that ND5 is involved in complex I assembly25. Furthermore, we found a decreased assembly of complex IV in cells with 98% of m.12955A > G mutation, but not in cells with 65% m.12955A > G mutation, which have relatively normal complex I assembly. Thus, it is more likely that the stability of complex IV is partially dependent on its assembly into a supercomplex containing complex I. A similar theory has been suggested by others and us. Indeed, in ours previous report, was found that complex IV was essential for the assembly and stability of complex I19.
Being the largest subunit of 13 mtDNA-encoded proteins, mutations in the ND5 subunit are frequently observed in patients with mitochondrial defects26. ND5 mutations have been reported in many types of mitochondrial diseases with a broad spectrum of clinical phenotypes ranging from mild to severe17. Diseases such as LHON27, adult encephalopathy11, MELAS28, myoclonic epilepsy with ragged red fibers (MERRF)29 and Leigh syndrome30 have been reported in patients carrying ND5 mutations. Clearly, the etiology of diseases related to ND5 mutations covers a broad spectrum and exact correlations between the site of mutation and the clinical outcome have yet to be defined17. The ND5 missense mutation 13371T > C, which is located in the third extramembranous domain facing the intermembrane space, has been identified in relation to muscular exercise intolerance but was not associated with other neurological abnormalities17. Interestingly, the patient with the 12955A > G mutation exhibited a similar clinical phenotype to that of the patient carrying the 13371T > C mutation, the only difference being that 12955A > G is located in the third extramembranous domain facing the mitochondrial matrix, while 13371T > C is located in the ninth transmembrane helix (location determined using http://www.uniprot.org/).
In summary, we report 12955A > G as a new missense mutation in the ND5 gene that is associated with exercise intolerance and developmental delay. Investigations of the underlying mechanism revealed that this mutation impaired mitochondrial oxidative phosphorylation which further increased cytotoxicity in a mutant load-dependent manner.
Materials and Methods
Mitochondrial respiratory complex enzymatic activity assay
Mitochondrial respiratory complex enzyme activities were measured in patient lymphocytes and cultured cells as described previously14,31.
Cell cultures
Transmitochondrial cybrids were obtained by fusion of mtDNA-less ρ0 human osteosarcoma 143B cells with platelets derived from the patient and her mother as described previously32. The transformant clones were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM; HyClone, Waltham, USA) containing 10% cosmic calf serum (Gibco, Carlsbad, USA).
mtDNA analysis
Genomic DNA was extracted using a sodium dodecyl sulfate (SDS) lysis protocol as described previously33. The entire mtDNA genome was Sanger sequenced using 24 previously reported pairs of mtDNA primers34. To quantify mutant loads, an allele specific amplification based real-time PCR method was used to determine the percentage of 12955G in total mtDNA. Real-time PCR reactions were performed on a Step-One plus Real-Time PCR system (Applied Biosystems, Foster City, USA) using the SYBR® Green qPCR Master Mix (Takara, Dalian, China). The sequences of the allele specific primers were as follows: mtDNA 12955A: forward, 5’-CAAATAGCCCTTCTAAACGCTA-3’; mtDNA 12955G: forward, 5’-CAAATAGCCCTTCTAAACGCTG-3’; reverse primer for mtDNA 12955A and mtDNA 12955G, 5’-CGCTGAGCCAGTCAGTGT-3’. The PCR amplification efficiencies of these primers were between 90% and 110%. The mutant load of 12955G was calculated using the following equation: Percentage of 12955G = 2(Ct12955A-Ct12955G)/(1 + 2(Ct12955A-Ct12955G)) × 100%. For the analysis of m.12955A > G mutations in healthy individuals, the primers listed below were used to sequence the ND5 gene: forward, 5’-AAACAACCCAGCTCTCCCTAA-3’; reverse, 5’- TCGATGATGTGGTCTTTGGA -3’.
Oxygen consumption
Endogenous oxygen consumption by intact cells was determined using a Clark-type oxygen electrode (Hansatech, Norfolk, United kingdom) as described previously35. After recording the basal respiration, oligomycin (2.5 μg/ml) (Sigma, St. Louis, USA) was added to measure the un-coupling respiration of the cells.
ATP measurements
ATP was measured using an ATP measurement kit (Molecular Probes, Carlsbad, USA) according to the manufacturer’s instructions. Briefly, cells were grown in 6-well plates to approximately 80% confluence. Approximately 1 × 106 cells were washed with cold phosphate buffered saline (PBS) buffer and then boiled in 100 μl Boiling Buffer (100 mM Tris, 4 mM EDTA, adjusted to pH 7.75 with acetic acid) for 90 s. Supernatants were retrieved by centrifugation at 10,000 × g for 1 min. ATP contents were determined by measuring the luminescence of supernatants mixed with Luciferase Assay buffer using a Varioskan™ Flash Multimode Reader (Thermo Scientific, Waltham, USA). ATP luminescence was normalized by protein concentration. To measure the generation of respiratory complex I-related ATP, a parallel set of cells was incubated with 200 nM rotenone (Sigma, St. Louis, USA) for 24 h before measuring the ATP.
Lactate and ROS measurement
The extracellular lactate level was measured using a fluorimetric-based lactate assay kit (Amplite, Foster city, USA) according to the manufacturer’s instructions. Briefly, culture medium was filtered through a 10 kDa molecular weight (MW) spin filter (Millipore, Darmstadt, Germany) and fluorescence was record using a Varioskan™ Flash Multimode Reader (Thermo Scientific, Waltham, USA).
Mitochondrial ROS were measured according to published protocols36. Briefly, cells were washed in Hank’s buffered salt solution (HBSS), resuspended in HBSS containing 5 μM Mito SOX (Molecular Probes, Carlsbad, USA) and incubated at 37°C for 5 min. Cells were then washed (twice) with HBSS and fluorescence was recorded using a Varioskan™ Flash Multimode Reader (Thermo Scientific, Waltham, USA). To measure ROS levels in the presence of rotenone, cells were incubated with 1000 nM rotenone for 1 h.
Mitochondrial protein preparation, blue native PAGE and in-gel activity assay
Mitochondria from cultured cells were isolated as previously described37. Mitochondrial membrane proteins were isolated from whole cells or mitochondria with n-dodecyl-β-D-maltoside (DDM, Sigma, St. Louis, USA) at the required ratio38. Protein (60 μg) containing 0.5% Blue G-250 (Sigma, St. Louis, USA) and 5% glycerol were separated by BN-PAGE (3–11% gel) as previously described38. For complex I in-gel activity assays, gels were soaked in assay buffer [25 mg nitrotetrazolium blue (NTB) (Sigma, St. Louis, USA) and 10 μl 1 mg/ml reduced nicotinamide adenine dinucleotide (NADH) (Sigma, St. Louis, USA) in 10 ml 5 mM Tris/HCL (pH 7.4)] for approximately 1 h. For complex IV assays, gels were soaked in Assay buffer [50 mM NaH2PO4 (pH 7.4), 5 mg 3, 3’-Damiobenzidine tetra hydrochloride hydrate (DAB) (Sigma, St. Louis, USA) and 50 μM cytochrome C (Sigma, St. Louis, USA)].
Immunoblotting
Proteins separated by BN-PAGE or SDS-PAGE were transferred to 0.22 μm PVDF membrane (Bio-Rad, Hercules, USA) using a semi-dry transfer system (15 V for 2 h, Bio-Rad, Hercules, USA). After blocking the membranes with 5% non-fat dried milk in Tris-buffered saline with Tween (TBST) [150 mM NaCl, 15 mM Tris-HCl (pH 7.5), 0.1% Tween 20] for approximately 2 h, proteins were probed using anti-NDUFA13 (1:1,000; MitoSciences, Eugene, USA) or anti-succinate dehydrogenase complex, subunit A (SDHA) (1:1,000; MitoSciences, Eugene, USA), anti-core2 (1:1,000; MitoSciences, Eugene, USA), anti-cytochrome c oxidase subunit IV (COXIV) (1:1,1000; MitoSciences, Eugene, USA), anti-ATP synthase subunit alpha (1:1,1000; MitoSciences, Eugene, USA), anti-voltage-dependent anion channel (VDAC) (1:1,000; Cell Signaling, Danvers, USA) antibodies and then incubated with an alkaline phosphatase-conjugated anti-mouse IgG (1:2,000; Cell Signaling, Danvers, USA) or horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000; Cell Signaling, Danvers, USA) secondary antibody. Membranes were washed with TBST after each antibody probing. Signals were detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, USA) or 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (BCIP/NBT) substrate (Promega, Madison, USA). Integrated optical density (IOD) quantification was performed using a Gel-Pro Analyzer 4.0 (MediaCybernetics, Warrendale, USA).
Statistical analysis
The data are presented as mean ± SD of three independent experiments. Statistical significance was evaluated by a one-way ANOVA or independent Student’s t test using SPSS 16.0 software (IBM, Armonk, USA). A null hypothesis was rejected when P <0.05.
Additional Information
How to cite this article: Fang, H. et al. Exercise intolerance and developmental delay associated with a novel mitochondrial ND5 mutation. Sci. Rep. 5, 10480; doi: 10.1038/srep10480 (2015).
References
Wallace, D. C. A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: a dawn for evolutionary medicine. Annual review of genetics 39, 359–407, 10.1146/annurev.genet.39.110304.095751 (2005).
Schon, E. A., DiMauro, S. & Hirano, M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nature reviews. Genetics 13, 878–890, 10.1038/nrg3275 (2012).
Nouws, J., Nijtmans, L. G., Smeitink, J. A. & Vogel, R. O. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain : a journal of neurology 135, 12–22, 10.1093/brain/awr261 (2012).
Skladal, D., Halliday, J. & Thorburn, D. R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain : a journal of neurology 126, 1905–1912, 10.1093/brain/awg170 (2003).
Hirst, J. Mitochondrial complex I. Annual review of biochemistry 82, 551–575, 10.1146/annurev-biochem-070511-103700 (2013).
Baradaran, R., Berrisford, J. M., Minhas, G. S. & Sazanov, L. A. Crystal structure of the entire respiratory complex I. Nature 494, 443–448, 10.1038/nature11871 (2013).
Perales-Clemente, E. et al. Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Molecular and cellular biology 30, 3038–3047, doi: 10.1128/MCB.00025-10 (2010).
Swalwell, H. et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. European journal of human genetics : EJHG 19, 769–775, 10.1038/ejhg.2011.18 (2011).
Lazarou, M., Thorburn, D. R., Ryan, M. T. & McKenzie, M. Assembly of mitochondrial complex I and defects in disease. Biochimica et biophysica acta 1793, 78–88, 10.1016/j.bbamcr.2008.04.015 (2009).
Wray, C. D. et al. A new mutation in MT-ND1 m.3928G > C p.V208L causes Leigh disease with infantile spasms. Mitochondrion 13, 656–661, 10.1016/j.mito.2013.09.004 (2013).
Malfatti, E. et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain : a journal of neurology 130, 1894–1904, 10.1093/brain/awm114 (2007).
Ugalde, C. et al. Impaired complex I assembly in a Leigh syndrome patient with a novel missense mutation in the ND6 gene. Annals of neurology 54, 665–669, 10.1002/ana.10734 (2003).
Kirby, D. M. et al. Mutations of the mitochondrial ND1 gene as a cause of MELAS. Journal of medical genetics 41, 784–789, doi: 10.1136/jmg.2004.020537 (2004).
Dunning, C. J. et al. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. The EMBO journal 26, 3227–3237, 10.1038/sj.emboj.7601748 (2007).
Spyropoulos, A. et al. Near-identical segregation of mtDNA heteroplasmy in blood, muscle, urinary epithelium and hair follicles in twins with optic atrophy, ptosis and intractable epilepsy. JAMA neurology 70, 1552–1555, 10.1001/jamaneurol.2013.4111 (2013).
Taylor, R. W., Taylor, G. A., Morris, C. M., Edwardson, J. M. & Turnbull, D. M. Diagnosis of mitochondrial disease: assessment of mitochondrial DNA heteroplasmy in blood. Biochemical and biophysical research communications 251, 883–887, 10.1006/bbrc.1998.9553 (1998).
Downham, E. et al. A novel mitochondrial ND5 (MTND5) gene mutation giving isolated exercise intolerance. Neuromuscular disorders: NMD 18, 310–314, 10.1016/j.nmd.2008.01.003 (2008).
Diaz, F., Fukui, H., Garcia, S. & Moraes, C. T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Molecular and cellular biology 26, 4872–4881 (2006).
Li, Y. F. et al. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. Journal of Biological Chemistry 282, 17557–17562 (2007).
Jones, R. P. O., Durose, L. J., Findlay, J. B. C. & Harrison, M. A. Defined sites of interaction between subunits E (Vma4p), C (Vma5p) and G (Vma10p) within the stator structure of the vacuolar H+-ATPase. Biochemistry 44, 3933–3941 (2005).
Gattermann, N. et al. Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 90, 4961–4972 (1997).
Perales-Clemente, E. et al. Five Entry Points of the Mitochondrially Encoded Subunits in Mammalian Complex I Assembly. Molecular and Cellular Biology 30, 3038–3047, 10.1128/Mcb.00025-10 (2010).
Andrews, B., Carroll, J., Ding, S., Fearnley, I. M. & Walker, J. E. Assembly factors for the membrane arm of human complex I. Proceedings of the National Academy of Sciences of the United States of America 110, 18934–18939, 10.1073/pnas.1319247110 (2013).
Bai, Y., Shakeley, R. M. & Attardi, G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol Cell Biol 20, 805–815 (2000).
Cardol, P. et al. In Chlamydomonas, the loss of ND5 subunit prevents the assembly of whole mitochondrial complex I and leads to the formation of a low abundant 700 kDa subcomplex. Biochimica et biophysica acta 1777, 388–396, 10.1016/j.bbabio.2008.01.001 (2008).
Blok, M. J. et al. Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J Med Genet 44, e74, 10.1136/jmg.2006.045716 (2007).
Mayorov, V., Biousse, V., Newman, N. J. & Brown, M. D. The role of the ND5 gene in LHON: characterization of a new, heteroplasmic LHON mutation. Ann Neurol 58, 807–811, 10.1002/ana.20669 (2005).
Corona, P. et al. A novel mtDNA mutation in the ND5 subunit of complex I in two MELAS patients. Ann Neurol 49, 106–110 (2001).
Naini, A. B. et al. Novel mitochondrial DNA ND5 mutation in a patient with clinical features of MELAS and MERRF. Arch Neurol 62, 473–476, 10.1001/archneur.62.3.473 (2005).
Shanske, S. et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol 65, 368–372, 10.1001/archneurol.2007.67 (2008).
Birch-Machin, M. A. & Turnbull, D. M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods in cell biology 65, 97–117 (2001).
Chomyn, A. et al. Platelet-mediated transformation of mtDNA-less human cells: analysis of phenotypic variability among clones from normal individuals–and complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. American journal of human genetics 54, 966–974 (1994).
Sambrook, J. & Russell, D. W. Molecular cloning: a laboratory manual. (Cold Spring Harbor Laboratory Press, 2001).
Rieder, M. J., Taylor, S. L., Tobe, V. O. & Nickerson, D. A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic acids research 26, 967–973 (1998).
Bai, Y. & Attardi, G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. The EMBO journal 17, 4848–4858, 10.1093/emboj/17.16.4848 (1998).
Park, J. S. et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet 18, 1578–1589, 10.1093/hmg/ddp069 (2009).
Fernandez-Vizarra, E. et al. Isolation of mitochondria for biogenetical studies: An update. Mitochondrion 10, 253–262, 10.1016/j.mito.2009.12.148 (2010).
Wittig, I., Braun, H. P. & Schagger, H. Blue native PAGE. Nature protocols 1, 418–428, 10.1038/nprot.2006.62 (2006).
Acknowledgements
This work was supported by grants from the Chinese National Science Foundation (81101506, 81471097), Zhejiang Provincial Natural Science Foundation of China (LQ15H070005) and the Start Foundation of Wenzhou Medical University (to Hezhi Fang).
Author information
Authors and Affiliations
Contributions
H.S., X.Y.L., Y.L.Y. and Y.Z. collected clinical data; J.X.L., H.Z.F., X.Y.L., Y.D., Y.Y.M., Y.P.L. and Y.L.Y. did the clinical evaluation; H.Z.F., H.S., D.Y.S., F.J.L., Y.D., B.L. and L.J.S. performed the experiments; J.X.L., H.Z.F., Y.L.Y. and Y.D.B. designed the experiment; J.X.L., H.Z.F., Y.L.Y. and H.S. wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fang, H., Shi, H., Li, X. et al. Exercise intolerance and developmental delay associated with a novel mitochondrial ND5 mutation. Sci Rep 5, 10480 (2015). https://doi.org/10.1038/srep10480
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10480
This article is cited by
-
Transcriptome analysis reveals the regulatory effects of Bacillus amyloliquefaciens and Bacillus pumilus on immune and digestive related genes in the spleen of weanling black goats
Functional & Integrative Genomics (2023)
-
A missense MT-ND5 mutation in differentiated Parkinson Disease cytoplasmic hybrid induces ROS-dependent DNA Damage Response amplified by DROSHA
Scientific Reports (2017)
-
Novel mutation of ND4 gene identified by targeted next-generation sequencing in patient with Leigh syndrome
Journal of Human Genetics (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
