
Abstract
PFAPA syndrome is the most common autoinflammatory syndrome in children from Western countries. In spite of its strong familial clustering, its genetic basis and inheritance pattern are still unknown. We performed a comprehensive genetic study on 68 individuals from 14 families. Linkage analysis suggested a susceptibility locus on chromosome 8, but direct molecular sequencing did not support this initial statistical finding. Exome sequencing revealed the absence of any gene that was mutated in all patients. Exhaustive screening of genes involved in other autoinflammatory syndromes or encoding components of the human inflammasome showed no DNA variants that could be linked to PFAPA molecular pathology. Among these, the previously-reported missense mutation V198M in the NLRP3 gene was clearly shown not to co-segregate with PFAPA. Our results on this relatively large cohort indicate that PFAPA syndrome is unlikely to be a monogenic condition. Moreover, none of the several genes known to be involved in inflammation or in autoinflammatory disorders seem to be relevant, alone, to its etiology, suggesting that PFAPA results from oligogenic or complex inheritance of variants in multiple disease genes and/or non-genetic factors.
Similar content being viewed by others

Introduction
Autoinflammatory syndromes (AIS) are a group of disorders characterized by attacks of inflammation that are not associated with the identification of autoreactive lymphocytes or of an external inflammation-triggering agent1. The majority of these syndromes are inherited as Mendelian traits, although a strong environmental influence has also been suggested for some forms2. Genes involved in AIS are linked to the inflammatory activation pathway and in particular to the activation of interleukin 1β (IL1β)3.
Periodic fever with aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome belongs to the AIS group, although the etiology of this disease is still unknown. Described for the first time by Marshall in 19874, it is characterized by high fever (higher than 39 °C) lasting three to seven days and reoccurring regularly every three to eight weeks, alongside with at least one of these other symptoms: aphthous stomatitis, pharyngitis, or cervical adenitis5. It is an early-onset disease (usually before the age of 5 years) and in general it completely resolves before adulthood. However, cases of PFAPA that were persistent after adolescence have also been reported6,7,8,9. Patients are asymptomatic between episodes and have no developmental or growth problems. During PFAPA febrile attacks, an increase of IL1β cytokines has been shown both at the gene10 and protein levels11, suggesting the involvement of IL1β release in the etiology of the disease.
The genetic origins of PFAPA are one of the most debated features of this syndrome. Although it is generally considered a sporadic disease12, familial clustering has been observed, suggesting the presence of a possible hereditary component13. It is not unusual to ascertain families having more than one member affected with PFAPA14,15 and positive family history for recurrent fevers can be detected in about half of PFAPA patients16. Several studies have investigated a possible involvement of the genes responsible for Familiar Mediterranean Fever (FMF, gene MEFV), TNF-Receptor Associated Periodic Syndrome (TRAPS, gene TNFRSF1A) Hyper IgD Syndrome (HIDS, gene MVK) and Cryopyrin Associated Periodic Syndrome (CAPS, gene NLRP3) in PFAPA cohorts11,17,18,19,20,21. An interesting hypothesis is that PFAPA could represent an unspecific, milder form of other AIS for which DNA variants in known AIS genes and present in the general population could be involved. An example is the NLRP3 variant V198M (or, more precisely, p.V200M, rs121908147), detected in the control population with an allele frequency of ~1% and reported as a causative mutation for CAPS22, as well as to be associated with milder diseases or with other unspecified AIS23,24,25. V198M was also identified in PFAPA patients, suggesting a possible role of NLRP3 hypomorphic alleles in the pathogenesis of the disease11, although a clear genotype-phenotype association is still missing. Recently, a heterozygous deletion of the gene SPAG7 consequent to a chromosomal translocation was reported in a single individual affected with PFAPA and displaying other syndromic features such as growth retardation, facial dysmorphisms and dysplastic brachymesophalangy26, However, these data could not be replicated and it is currently not clear whether the link between SPAG7 and PFAPA is causal or coincidental.
In this work, we perform for the first time an unbiased, comprehensive genetic analysis on a collection of families with PFAPA syndrome. Our data indicate that PFAPA is neither a monogenic disease nor a condition that can be clearly associated with DNA changes in other AIS genes. In addition, we demonstrate that the NLRP3 DNA variant V198M is not associated with this disease.
Results
PFAPA syndrome shows an apparent autosomal dominant with incomplete penetrance pattern of inheritance, if considered as a Mendelian disease
We analyzed 68 individuals from 14 families segregating PFAPA by genome-wide SNP genotyping, whole-exome sequencing, or both. Pedigree analysis suggested clear inheritance, compatible with an autosomal dominant with incomplete penetrance model, if Mendelian inheritance with no genetic heterogeneity is assumed as a model (Fig. 1). Because of the lack of genetic data for this disease to infer the penetrance factor from the literature, we empirically set it to be around 50% following pedigree examination. Following these observations, we decided to perform a genome-wide linkage analysis to identify a possible locus carrying the causative gene of PFAPA syndrome. We selected a subset of informative families (families A, B, C, H, K, L, O) based on their clinical information. Multifamily whole genome analysis considering non-parametric methods revealed a single and unique peak on chromosome 8 with a maximum LOD score of 2.9 (Fig. 2a). This peak was also conserved with a parametric method by considering a fully penetrant inheritance pattern with a LOD score of 3.4 (Fig. 2b). The identified region was of 11 Mb and spanned bands 8q21.1 to 8q24.4 of chromosome 8 (Fig. 2c). This region is scarcely populated, as it contains only 40 RefSeq genes, of which only 32 were protein coding (Fig. 2d – and Supplementary Table 1). We selected 3 affected individuals (B1, L1 and C2) to be screened for all exons of all genes that are present in the interval. Analysis of DNA variants failed to reveal any single gene that was mutated in all three affected individuals within the identified region, with the exception of two non-coding variants representing common polymorphisms (Supplementary Table 2).

Families analyzed
Alphanumeric codes indicate individuals who were investigated by genome-wide SNP genotyping, while arrows indicate individuals whose DNA was processed by exome sequencing (some individuals were analyzed by both techniques). Boxes indicate families that underwent linkage studies.

Linkage analysis of PFAPA families
a) Output of Merlin linkage analysis for linear (black line) and exponential (blue line) non-parametric models. For our study, the exponential model gives a better resolution, since it is indicated for a small number of families and large allelic sharing. b) Output for a parametric model (autosomal dominant with incomplete penetrance). The peak at chromosome 8 is still conserved. c) Location of the linkage interval on the long arm of chromosome 8, between markers rs221160 and rs221962. d) Protein coding genes present in the identified interval.
Exome capture and NGS show no common disease gene associated with PFAPA
Despite the negative results from the screening of the chromosome interval, we could not completely exclude a priori a monogenic model of inheritance. Therefore we decided to select some key individuals to be whole-exome sequenced. Inclusion criteria were the availability and the quality of the extracted DNA, as well as the size and the quality of the clinical information regarding the family (Table 1). We sequenced, whenever possible, two affected individuals within the same family, or one obligated carrier and a patient, for a total of 11 disease-carrying subjects and one unaffected family member (Fig. 1, arrows).
Under the hypothesis that PFAPA is a monogenic condition with no genetic heterogeneity, we scanned the list of DNA variants present in all patients, in order to identify a gene carrying rare variants (minor allele frequency, or MAF < 2%) in more than 90% of the analyzed samples (to allow the presence of false negatives). In total, we detected 14 genes that satisfied these criteria (Table 2). Four of them belonged to the MUC family and represented false positives results, similar to those obtained by other exome sequencing projects27. The same was true for mutations in six other genes (WNK1, ZNF384, TTN, GOLGAL6L1, C11orf40 and KIAA1751) that, for various reasons, could be recognized as sequencing artifacts28. The remaining four genes (NBPF15, OR4C45, ZNF492 and PCMTD1) had many novel variants, which were however also present in controls. No common gene carrying rare variants that could be compatible with pathogenic mutations could therefore be identified in our cohort.
No rare or potentially abnormal DNA variants were found in the SPAG7 gene, in any of the patients analyzed.
Screening for mutations in genes for autoinflammatory recurrent fever conditions does not support their involvement in PFAPA etiology
Since the presence of rare variants in other inflammatory genes could have an impact on the PFAFA phenotype, we decided to search specifically for variants in all known autoinflammatory genes in our cohort (Supplementary Table 3). Screening of the MEFV gene, associated with FMF, revealed the presence of polymorphisms in heterozygous state with unknown function for patients O1 (p.I591T, rs11466045, MAF 0.07) and R1 (p.F425Y, rs11466045, MAF 0.001). None of these were present in the other analyzed members of the family. Conversely, in family A, we identified a variant (p.E148Q, rs3732930, MAF 0.06) shared among the two affected individuals. Interestingly, both patients of family A also carried a heterozygous change in NLRP3 related to CAPS (p.Q703K, rs35829419, MAF 0.04). In family B, we identified a rare variant in the gene TNFRSF1A, usually associated with milder forms of TRAPS (p.R121Q, rs4149584, MAF 0.018). This change cosegregated with the disease. We also identified a novel change in the gene NLRP12, associated with familial cold autoinflammatory syndrome-2, in patient R1 (p.R211C). This change, located in a well-conserved region of the protein (Table 3), was also present in R1’s unaffected sister.
The NLRP3 variant p.V198M associated with CAPS does not cosegregate with PFAPA affected status
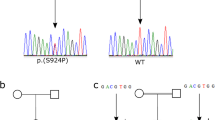
Analysis of NLRP3 in family O revealed the presence of the variant V198M (p.V200M, rs12908147, MAF 0.01), often associated with milder forms of CAPS23,29. This variant was found in heterozygous state only in the obligated carrier member O5, whereas it was not present in the proband. Cosegregation analysis on available members of the family revealed that this change is present also in the two healthy siblings and it is absent in the affected one, confirming that this change is not involved in the etiology of PFAPA in this family (Fig. 3).

Cosegregation analysis for the V198M variant in NLRP3, in family O.
Variant V198M (p.V200M) in gene NLRP3 does not cosegregate with PFAPA in this pedigree (healthy members carry it, whereas the affected member does not), suggesting that this polymorphism is not associated with the disease.
Several rare variants with uncertain function were present in inflammasome-related genes
Since an impairment in IL-1β and NF-κB signaling and production has been reported in individuals with PFAPA syndrome10, we decided to look closely at all known genes belonging to the inflammasome complex, with particular attention to genes involved in the production of mature IL-1β. All identified variants in these genes are listed in Table 3. We identified two non-annotated changes in NLRP5, for patients 2940 and F1 (p.R274Q and p.N255S, respectively). Again, patient 2940 presented another rare variant in the gene NLRP2, p.L607P (rs182098487, MAF 0.001). A novel change, p.G504D, was found in this same gene in patient B7, although it was not found in the affected cousin. Among the Nod-Like Receptor genes, NLRP2 showed the highest number of variants within PFAPA subjects, but these were also rather common in the control CoLaus Lausanne cohort (p.S4L, rs142463014, for 2492, MAF 0.09; p.I331V, rs61735077, for R1 and R2, MAF 0.05). Another common variant in NLRP4 was p.T162M (rs117212164, MAF 0.03), co-segregating with the condition in family O and affecting a conserved residue in the NACHT domain of the NLRP4 protein. Among other genes from the NLR families we identified a rare change in NLRP10, p.I384T (rs150112481, MAF 0.01) although only in patient O1 and the change p.S1025L (rs11671248, MAF 0.08) in NLRP11 in patient 2940. Not many rare variants were identified in the coding region of the NLRP1 gene, with the only exception being p.V939M (rs61754791, MAF 0.01), found only in patient 2942. Interestingly, however, both patient 2941 and family R shared a rare haplotype composed by different missense polymorphisms located at the C-terminal of the protein (Fig. 4). A similar haplotype has been recently associated with an impairment of IL-1β processing by the inflammasome. Common polymorphisms in CARD8 were found in a heterozygous state in our cohort and these have been previously associated with other defects of the inflammatory system. These polymorphisms were p.C10X (rs2043211, MAF 0.30) found in patients A1, B7, F1, 2940 and p.V148fs (rs140826611, MAF 0.06) in A2, F1, 2941 and 2942. Among other inflammasome genes, we identified a heterozygous missense change in NLRC4, better known as IPAF, p.G786V (rs149451729, MAF 0.03) in patient 2941; a novel change in NOD1 (p.C241G) in patient 2942, again in the NACHT domain of this protein. A summary of all analyzed variants identified in inflammasome genes and their affected functional domain is reported (Table 3).

Graphical representation of the haplotype identified on NLRP1 for two subjects.
The gene structure is depicted in the upper part. Amino acid changes identified in these individuals are reported. All these changes have a MAF of <0.1. PYD: pyrin domain; NACHT: Conserved NAIP, CIITA, HET-E and TP-1 domains; LRR: leucine rich receptor domain; CARD: Caspase activator and recruitment domain.
Discussion
PFAPA is a common auto-inflammatory disease with an overall good prognosis. Regular PFAPA fever flares have a significant impact on the quality of life of the patients and their families and are interfering with regular school attendance and normal daily activities. The delay to diagnosis may represent several months or years and children may be exposed to unnecessary diagnostic procedures. A better understanding of the pathogenesis of this disease would lead to more specific diagnostic tools and might relieve the suffering of patients and their families. Clearly the pathogenesis of the disease is linked to the immune system, but no convincing information about the etiology and in particular about the genetic basis of PFAPA has been obtained so far.
In our clinical practice, we identified a number of families for which PFAPA seems to have a genetic basis. Based on pedigrees analysis, we could infer an autosomal dominant model of inheritance, with a penetrance factor of almost 50%. However, some families showed an apparently dominant inheritance with full penetrance (e.g. family K), whereas for others a recessive status could not be excluded a priori (e.g. family F). According to these observations, we selected the seven most informative families of our collection to perform a genome-wide linkage study. This analysis revealed a clear peak on the long arm of chromosome 8, notably between 8q24.1 and 8q24.3. This region contained a small number of genes and was very close to the telomere of chromosome 8. The significance of the identified area was confirmed using non-parametric analysis and by comparing haplotypes of affected individuals against non-affected ones. Interestingly, the identified region contained several lncRNAs and many miRNAs, as well as other unknown genes or genes without a clear function. Based on these data we therefore performed a custom sequence capture of all known exons for genes in the interval. The custom capture chip was designed based on annotation of build hg35.1 of the human genome. This implies that we could have missed some recently discovered genes and all the hypothetical genes and lncRNAs that corresponded to the majority of elements annotated in the locus. The sequencing did not reveal any single mutated gene shared by all affected individuals.
To exclude the possibility that we did not detect a causative disease gene because of inconsistencies in the inferred inheritance pattern, we decided to sequence the entire exome of 11 affected individuals, one “obligate carrier” (i.e. a healthy individual connecting two branches of a pedigree with affected subjects) and one unaffected family member. Our analyses revealed the absence of a unique gene that was mutated in all the affected individuals, including genes and noncoding RNAs lying in the critical region on chromosome 8. Although we cannot exclude a priori that mutations that escape exome sequencing such as intronic variants and copy number variations could be the cause of the disease, our data disfavor genetic homogeneity for PFAPA.
We then decided to investigate the possibility that mild mutations or rare variants in other known autoinflammatory genes could cause the PFAPA phenotype. This hypothesis is currently a matter of debate, since the diagnosis of milder forms of autoinflammatory disease and PFAPA is often overlapping, leading to diagnostic uncertainties. In particular, low-frequency polymorphisms (common in 1-4% of the healthy population) have been associated with PFAPA or milder forms of autoinflammatory diseases, mostly because no other strong changes in candidate genes were identified17,19,30. Our analysis revealed that, although we identified a few rare missense changes in some inflammatory genes, the frequencies of these variants were not different from those that were present in the general population, indicating that they may not be sufficient to induce the disease. Moreover, some of these changes did not co-segregate with the phenotype, or were also present in healthy members of the family. This was for example observed for the NLRP3 variant V198M (p.V200M). This change has been reported in several different forms of NLRP3-associated syndrome, such as classical CAPS22,23,29, or other autoinflammatory manifestation24,31, although it is classified as a low penetrant mutation, due to the fact that it is present in a number of healthy carriers (in the Lausanne control population it is present with a frequency of about 1%). The role of this variant is still debated, but it is regrettably reported as a bona fide pathogenic mutation in medical genetics diagnostic protocols. The presence of this variant only in the unaffected individuals of family O strongly contradicts its possible role in generating an inflammatory phenotype. A recent paper, probably the most complete clinical study of V198M, showed that 19 cases of CAPS out of 830 carried V198M, although 3 of these harbored other possible candidates in other inflammatory genes and V198M was present in 7 asymptomatic individuals related to these 19 patients25. Furthermore, Valine at codon 200 is not at all conserved through evolution, suggesting that this amino acid residue is likely not detrimental for protein function. Other mammals such as rat, dog and mouse carry a methionine at the same position of their natural NLRP3 orthologues. Altogether, these data indicate that V198M (p.V200M) is likely a neutral human polymorphism and that patients carrying this variant should probably be re-analyzed for other causative mutations.
Another example is the novel change found in patient R1, the NLRP12 p.R211C, which is present in a well-conserved domain and has a predicted strong impact on protein function. Recently a similar missense mutation (p.R352C) in the same NACHT domain of this gene was associated with genetically unexplained periodic fevers32, with a PFAPA-like phenotype and was shown to cause a functional defect in Caspase-1 signaling. The fact that R1’s unaffected sister carries this allele likely indicates that this change has no effects (or has at best a hypomorphic role) at the clinical level. It is interesting to notice that the affected patient (R1) also carried another very rare change in the gene MEFV (p.F425Y) with function unknown. The intriguing hypothesis of digenic or multigenic mutations causing the impairment of the inflammasome seemed to be reinforced by the identification of two polymorphisms often associated with the disease in family A. More specifically, we identified a change in the MEFV gene (p.E148Q) and in NLRP3 (p.Q703K), both common polymorphisms with a possible functional effect on the protein. In particular, for p.Q703K, functional impairment of NLRP3 has been clearly demonstrated33. Unfortunately this digenic pattern of inheritance was not identified in other affected subjects within known autoinflammatory-associated genes34. Nevertheless we cannot exclude, based on this observation, that other rare variants present in other inflammasome-associated genes could be involved in the etiology of PFAPA.
To investigate this possibility we carefully screened some of the genes that compose the inflammasome set (in particular the NLR family members), based on current literature35,36,37. We identified a few novel variants in NLRP2, NLRP5 and NOD1, as well as some very rare variants in other inflammasome-associated genes, such as NLRP10, NLRP4, NLRP11 and NLRC4. Interestingly, the majority of these genes were involved in IL-1β inflammasome activation, or in the NF-κB pathway. For example, new evidence demonstrated the high similarity and redundancy across the structure of NLRP1, NLRP6, NLRP10, NLRP3 and NLRP1238. Although the function within the inflammasome for many of these members has not yet been clarified, structural similarities among NLRs suggest that all these components could somehow mediate inflammation in response to different stimuli and trigger IL-1β production37. Moreover, we identified two samples carrying a specific rare haplotype in NLRP1 that has recently been associated with vitiligo and autoimmune diseases and connected to an increase of IL-1β release in patients carrying the haplotype39. Dominant mutations in the same gene have been shown to cause systemic inflammation in mouse models40. Another observation from the analysis on the possible effect on protein structure was that the majority of identified variants seemed to be present in or in proximity of the NACHT domain. This domain has an ATPase activity and is crucial for the self-oligomerization (usually to form heptamers or hexamers) of NLRs to form the structure necessary for inflammasome assembly41. NACHT domain mutations have been associated with structural changes that lead to a continuous activation of the inflammasome complex. A link between mutations affecting NACHT and different autoinflammatory diseases has been observed in NLRP3 and NOD1 for example42,43, reinforcing the hypothesis of a gain-of-function effect of these mutations on the formation of the complex.
It is interesting to observe that every single affected individual in this cohort presented more than one rare variant in one of the inflammasome-composing genes. Unfortunately, due to large number of genes involved, it is impossible to perform a meaningful statistical analysis based on this observation, unless extremely large groups of PFAPA patients are recruited and analyzed, possibly as part of an extended international effort. However, it may not be unreasonable to start thinking of a “total inflammatory burden” similar to the concept of “total ciliary burden” proposed for disease such as the Bardet-Biedl syndrome, that has developed from a simple Mendelian disorder into a trigenic44,45 and finally into an oligogenic disorder46.
In conclusion, the absence of a common gene harboring exonic mutations in all the affected individuals analyzed in this work suggests that PFAPA syndrome is transmitted either as Mendelian disease with high genetic heterogeneity, or as an oligogenic or complex trait. Mutations in noncoding or poorly covered regions of the genome that may have escaped our analysis are also possible and further studies are needed to assess their potential presence in PFAPA patients. Oligogenic inheritance would be consistent with our observation that affected individuals may harbor multiple variants in inflammasome-associated genes. However, given the fact that such variants may be shared with a significant number of the general unaffected population, specific studies are needed to underpin or refute this hypothesis and extreme caution should be used prior to indicating causality.
Methods
Patient selection and ethical commitment
The families participating in this study were from a large collection of PFAPA patients who were ascertained at the Center for Pediatric Rheumatology of Western Switzerland at the Lausanne University Hospital and the Geneva University Hospital. The familial clustering and inheritance patterns were investigated by administrating a specific survey to family members of the affected patients. The diagnosis of PFAPA in family members was based on the report, by the affected persons or their parents, of a stereotypical and recurrent pattern of illness during their own childhood. The PFAPA phenotype was defined according to previously published clinical criteria47; in addition a genetic screening for one or more monogenic periodic fever syndromes (FMF, TRAPS, HIDS) was performed for selected individuals. Our study was designed in accordance with the tenets of the declaration on Helsinki and was approved by the Institutional Review Boards of the University of Lausanne and of the Lausanne University Hospital. Written informed consent was obtained from all patients who enrolled in the study. As controls for the screening of rare variants, data obtained by exome sequencing of 416 healthy anonymous individuals from a large population-based study done in Lausanne (the CoLaus cohort) were used48.
DNA extraction and lymphocytes immortalization
DNA was extracted from peripheral leukocytes (PBLs) of participants by using the NucleonBaCC3 DNA extraction kit (GE healthcare, Life Science). For some family members and for all probands, white mononucleate cells were isolated using Ficoll-Paque PLUS (GE healthcare Life Science), according to the manufacturer’s instruction. Once isolated, cells were processed for EBV immortalization using classical protocols49, optimized for our requirements as follows. After three washes with PBS and 2% FCS, 1 × 106 PBL cells were transferred to a 10 ml Falcon tube, diluted in 1 ml of LCC medium (RPMI 1640 medium, 20% serum, 2 μg/ml Cyclosporine A) and incubated with a EBV virus suspension for 1 h. Cells were then plated into single wells from a 12-well plate and kept in culture for ~1 month. Following this immortalization process, cells were frozen in RPMI 1640 with 20% serum and 10% DMSO for cryopreservation.
Genotyping and linkage analysis
Six out of 14 families were selected for whole genome genotyping, based on the availability of clinical information and the size of the family trees. DNA from all available members of each of these families was assessed by the Illumina Human Linkage12 array (Illumina, CA), containing more than 6000 highly heterozygous SNPs, with an average distance between SNPs of about 441 kb. Output was then analyzed using the Genotype Studio software (Illumina Inc., CA) and the SNPs calls were filtered based on quality and the presence of Mendelian inconsistencies. Genotype data were handled using the commercial software Progeny 7 (Progeny Software LLC) to generate appropriates files for linkage analysis software, carried out with the Merlin package and by using the Vital-IT high performance computer structure (the Swiss Institute of Bioinformatics, Lausanne, www.vital-it.ch). Both parametric and not-parametric analysis models were tested. Because of the lack of data on genetic background for this disease, we could not perform a classical computational approach to estimate the penetrance factor for the parametric analysis. Therefore, penetrance factor for parametric analysis was empirically calculated based on the observed families as a ratio between the number of affected patients and the total number of subjects predicted to be affected based on the model.
Next generation sequencing
In the first sequencing experiment, aimed at determining DNA variants on chromosome 8 interval, a custom made sequence capture chip, based on Agilent SureSelect technology, was designed by Genotypic (Bangalore, India). Captured DNA was then sequenced by Fasteris (Geneva, Switzerland) by using a single lane of the Illumina Genome Analyzer II (Illumina, San Diego CA).
For exomes, the entire capturing and sequencing procedures were performed at the Lausanne Genomic Facility (GTF). Genomic DNA samples were barcoded and exons were captured using the SureSelect Exome kit V5 (Agilent, Santa Clara, CA) according to the manufacturer’s protocols. Sequencing was performed using again a single lane of the Illumina Genome Analyzer II (Illumina, San Diego, CA). Reads were aligned to the reference sequence (human genome reference 19 – hg19) using Novoalign 2.08 (Novocraft.com). The obtained coverage was 10x or higher for 92% of the targeted regions. Base quality score recalibration, indel realignment, duplicates removal and SNP and INDEL calling were performed by using Genome Analysis Tools kit (GATK)50. Variant annotation and filtering was performed with both Annovar51 and the Ingenuity Variant Analyzer web interface (Ingenuity System, CA). A subset of the identified variants were confirmed and analyzed by classical Sanger sequencing according standard protocols. Statistical analysis on rare variants present in inflammasome related genes was performed using the Graphpad on-line calculator (http://graphpad.com). A two-tails χ2 test adjusted for 2 × 2 contingency table was used.
Additional Information
How to cite this article: Di Gioia, S. A. et al. Analysis of the genetic basis of periodic fever with aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome. Sci. Rep. 5, 10200; doi: 10.1038/srep10200 (2015).
References
Touitou, I. & Kone-Paut, I. Autoinflammatory diseases. Best Pract. Res. Clin. Rheumatol. 22, 811–829 (2008).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Masters, S. L., Simon, A., Aksentijevich, I. & Kastner, D. L. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu. Rev. Immunol. 27, 621–668 (2009).
Marshall, G. S., Edwards, K. M., Butler, J. & Lawton, A. R. Syndrome of periodic fever, pharyngitis and aphthous stomatitis. J. Pediatr. 110, 43–46 (1987).
Padeh, S. et al. Periodic fever, aphthous stomatitis, pharyngitis and adenopathy syndrome: clinical characteristics and outcome. J. Pediatr. 135, 98–101 (1999).
Padeh, S., Stoffman, N. & Berkun, Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr. Med. Assoc. J. 10, 358–360 (2008).
Cantarini, L., Vitale, A., Galeazzi, M. & Frediani, B. A case of resistant adult-onset periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome responsive to anakinra. Clin. Exp. Rheumatol. 30, 593 (2012).
Colotto, M. et al. PFAPA syndrome in a young adult with a history of tonsillectomy. Intern. Med. 50, 223–225 (2011).
Cazzato, M., Neri, R., Possemato, N., Puccini, R. & Bombardieri, S. A case of adult periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome associated with endocapillary proliferative glomerulonephritis. Clin. Rheumatol. 32 (Suppl 1) 33–36 (2013).
Stojanov, S. et al. Periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc. Natl. Acad. Sci. USA 108, 7148–7153 (2011).
Kolly, L. et al. Periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome is linked to dysregulated monocyte IL-1beta production. J. Allergy Clin. Immunol. 131, 1635–1643 (2013).
Hofer, M., Mahlaoui, N. & Prieur, A. M. A child with a systemic febrile illness - differential diagnosis and management. Best Pract. Res. Clin. Rheumatol. 20, 627–640 (2006).
Akelma, A. Z. et al. Is PFAPA syndrome really a sporadic disorder or is it genetic? Med. Hypotheses 81, 279–281 (2013).
Sampaio, I. C., Rodrigo, M. J. & Monteiro Marques, J. G. Two siblings with periodic fever, aphthous stomatitis, pharyngitis, adenitis (PFAPA) syndrome. The Pediatric infectious disease journal 28, 254–255 (2009).
Valenzuela, P. M., Majerson, D., Tapia, J. L. & Talesnik, E. Syndrome of periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) in siblings. Clin. Rheumatol. 28, 1235–1237 (2009).
Cochard, M. et al. PFAPA syndrome is not a sporadic disease. Rheumatology 49, 1984–1987 (2010).
Dagan, E., Gershoni-Baruch, R., Khatib, I., Mori, A. & Brik, R. MEFV, TNF1rA, CARD15 and NLRP3 mutation analysis in PFAPA. Rheumatol. Int. 30, 633–636 (2010).
Kovacs, L. et al. Elevated immunoglobulin D levels in children with PFAPA syndrome. Neuro. Endocrinol. Lett. 31, 743–746 (2010).
Gattorno, M. et al. Differentiating PFAPA syndrome from monogenic periodic fevers. Pediatrics 124, e721–728 (2009).
Toplak, N. et al. An international registry on autoinflammatory diseases: the Eurofever experience. Annals of the rheumatic diseases 71, 1177–1182 (2012).
Taniuchi, S. et al. MEFV Variants in Patients with PFAPA Syndrome in Japan. Open Rheumatol. J. 7, 22–25 (2013).
Aganna, E. et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness and AA amyloidosis. Arthritis Rheum. 46, 2445–2452 (2002).
Porksen, G. et al. Periodic fever, mild arthralgias and reversible moderate and severe organ inflammation associated with the V198M mutation in the CIAS1 gene in three German patients - expanding phenotype of CIAS1 related autoinflammatory syndrome. Eur. J. Haematol. 73, 123–127 (2004).
Loock, J. et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. The Journal of allergy and clinical immunology 125, 500–502 (2010).
Rowczenio, D. M. et al. Clinical characteristics in subjects with NLRP3 V198M diagnosed at a single UK center and a review of the literature. Arthritis Res. Ther. 15, R30 (2013).
Bens, S. et al. SPAG7 is a candidate gene for the periodic fever, aphthous stomatitis, pharyngitis and adenopathy (PFAPA) syndrome. Genes. Immun. 15, 190–194 (2014).
Fuentes Fajardo, K. V. et al. Detecting false-positive signals in exome sequencing. Human mutation 33, 609–613 (2012).
Musumeci, L. et al. Single nucleotide differences (SNDs) in the dbSNP database may lead to errors in genotyping and haplotyping studies. Hum. Mutat. 31, 67–73 (2010).
Hoffman, H. M., Mueller, J. L., Broide, D. H., Wanderer, A. A. & Kolodner, R. D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nature genetics 29, 301–305 (2001).
Pelagatti, M. A. et al. Long-term clinical profile of children with the low-penetrance R92Q mutation of the TNFRSF1A gene. Arthritis Rheum. 63, 1141–1150 (2011).
Touitou, I., Perez, C., Dumont, B., Federici, L. & Jorgensen, C. Refractory auto-inflammatory syndrome associated with digenic transmission of low-penetrance tumour necrosis factor receptor-associated periodic syndrome and cryopyrin-associated periodic syndrome mutations. Annals of the rheumatic diseases 65, 1530–1531 (2006).
Jeru, I. et al. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum. 63, 1459–1464 (2011).
Verma, D. et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1beta and IL-18 production. PloS one 7, e34977 (2012).
Touitou, I. et al. Infevers: An evolving mutation database for auto-inflammatory syndromes. Human mutation 24, 194–198 (2004).
Zambetti, L. P., Laudisi, F., Licandro, G., Ricciardi-Castagnoli, P. & Mortellaro, A. The rhapsody of NLRPs: master players of inflammation and a lot more. Immunol. Res. 53, 78–90 (2012).
Correa, R. G., Milutinovic, S. & Reed, J. C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Bioscience reports 32, 597–608 (2012).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Anderson, J. P. et al. Structural, expression and evolutionary analysis of mouse CIAS1. Gene. 338, 25–34 (2004).
Levandowski, C. B. et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proceedings of the National Academy of Sciences of the United States of America 110, 2952–2956 (2013).
Masters, S. L. et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37, 1009–1023 (2012).
Proell, M., Riedl, S. J., Fritz, J. H., Rojas, A. M. & Schwarzenbacher, R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PloS one 3, e2119 (2008).
Albrecht, M., Domingues, F. S., Schreiber, S. & Lengauer, T. Structural localization of disease-associated sequence variations in the NACHT and LRR domains of PYPAF1 and NOD2. FEBS Lett. 554, 520–528 (2003).
Aksentijevich, I. et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 56, 1273–1285 (2007).
Katsanis, N. et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293, 2256–2259 (2001).
Eichers, E. R., Lewis, R. A., Katsanis, N. & Lupski, J. R. Triallelic inheritance: a bridge between Mendelian and multifactorial traits. Ann. Med. 36, 262–272 (2004).
Davis, E. E. & Katsanis, N. The ciliopathies: a transitional model into systems biology of human genetic disease. Curr. Opin. Genet. Dev. 22, 290–303 (2012).
Thomas, K. T., Feder, H. M., Jr., Lawton, A. R. & Edwards, K. M. Periodic fever syndrome in children. The Journal of pediatrics 135, 15–21 (1999).
Firmann, M. et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc. Disord. 8, 6 (2008).
Brickell, P. M. Immortalization of human B-lymphocytes by epstein-barr virus. Methods in molecular biology 8, 213–218 (1992).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164 (2010).
Acknowledgements
This work was supported by the Swiss National Science Foundation (Grant 310030_138346), the Gebert Rüf Foundation, Switzerland (Rare Diseases - New Technologies grant) and the Novartis Foundation.
Author information
Authors and Affiliations
Contributions
SADG, ASF, MH, CR wrote the manuscript, SADG, NB, AvSG, FV, MH performed data acquisition, SADG, NB, ASF, MH, CR analyzed the data and SADG, ASF, MH, CR designed the study, All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gioia, S., Bedoni, N., von Scheven-Gête, A. et al. Analysis of the genetic basis of periodic fever with aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome. Sci Rep 5, 10200 (2015). https://doi.org/10.1038/srep10200
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10200
This article is cited by
-
NLRP3 gene variants and serum NLRP3 levels in periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome
Clinical Rheumatology (2023)
-
Synchronous disease onset and flares in siblings with PFAPA
Pediatric Rheumatology (2022)
-
The role of inflammatory mediators in the pathogenesis of periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome
Molecular Biology Reports (2022)
-
PFAPA (periodic fever, aphthous stomatitis, pharyngitis, and adenitis) syndrome: an overview of genetic background
Clinical Rheumatology (2021)
-
IL-1β blockade in periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome: case-based review
Rheumatology International (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
