
Abstract
The independent dietary shift from carnivore to herbivore with over 90% being bambooin the giant and the red pandas is of great interests to biologists. Althoughprevious studies have shown convergent evolution of the giant and the red pandas atboth morphological and molecular level, the evolution of the gut microbiota in thesepandas remains largely unknown. The goal of this study was to determine whether thegut microbiota of the pandas converged due to the same diet, or diverged. Wecharacterized the fecal microbiota from these two species by pyrosequencing the 16SV1–V3 hypervariable regions using the 454 GS FLX Titanium platform. We alsoincluded fecal samples from Asian black bears, a species phylogenetically closer tothe giant panda, in our analyses. By analyzing the microbiota from these 3 speciesand those from other carnivores reported previously, we found the gut microbiotas ofthe giant pandas are distinct from those of the red pandas and clustered closer tothose of the black bears. Our data suggests the divergent evolution of the gutmicrobiota in the pandas.
Similar content being viewed by others

Introduction
The advent of the high throughput next generation sequencing has allowed scientists toexplore the gut microbiota with an unprecedented depth. The evolution of the gutmicrobiota has recently received great interests1,2,3. Several factorssuch as diet and phylogeny have been reported to play important roles in shaping the gutmicrobiota at different taxonomic scales2,3,4,5,6,7.
The red and the giant pandas are interesting models to study the evolution of the gutmicrobiota as they are carnivores by phylogeny but herbivores by diet. Both speciesexperienced a dietary switch from carnivores to highly specialized bamboo eaters. Theyboth independently developed several similar morphological features such as the falsethumb8 in adaptation to the same dietary switch to bamboo. However,it is still unclear whether the gut microbiota in the red and the giant pandas convergeddue to the similar, highly specialized bamboo diet, or diverged corresponding to otherunknown factors.
To determine their evolutionary patterns we characterized the gut microbiotas in the twotypes of pandas and compared them to the gut microbiotas of black bears. We found adivergent evolution pattern in the gut microbiotas of the pandas.
Results
Comparison of the gut microbiota of the three carnivores
We characterized the gut microbiotas of 6 red and 5 giant pandas by sequencingthe 16S V1–V3 hyper variable region of their feces collected from thezoo. We also sequenced the gut microbiotas of 6 Asian black bears, which arephylogenetically closer to the giant panda than to the red panda. The sequenceswere processed and analyzed by using the mothur software package9. We retained a total of 63, 944 high quality reads after denoising, with anaverage of 3,761 sequences per sample ranging from 1,214 to 7,450. Thesesequences were assigned to 235 operational taxonomic units (OTUs). Sequencenumber for each sample was normalized to 1,200 by randomly subsampling tominimize the biases generated by sequencing depth. Theaverage ± SD Good’s coverage was99.3 ± 0.6% (TableS1).
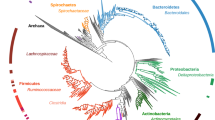
We examined the relationship between the gut microbiotas from different speciesby using Bray-Curtis distances10, which were visualized by adendrogram. Each branch on the tree represents one gut microbiota (Fig. 1). Interestingly, the gut microbiotas of the giantpandas located on different branches from those of the red pandas and clusteredcloser to those of the black bears (Fig. 1).

Clustering analysis of the evolution ofthe gut microbiotas of the black bears, the giant and the redpandas.
Gut microbiota trees were generated using theUnweighted Pair Group Method with Arithmetic Mean algorithm based on theBray-Curtis distances generated by mothur.
We also used principal coordinate analyses (PCoA) to examine the relationshipsbetween gut microbiota of the red and the giant pandas (Fig.2). We observed similar clustering patterns. On the PCoA plot, eachsymbol represents one gut microbiota. Consistent with the dendrograms, the gutmicrobiotas of the giant pandas were distinct from those of the red pandas.Analysis of molecular variance (AMOVA) of the Bray-Curtis distances proved thatthe differences between the gut microbiotas of the giant and the red pandas werestatistically significant (AMOVA, P < 0.05). Interestingly,the gut microbiota of the giant pandas clustered closer to the gut microbiota ofthe black bears than to the red pandas, consistent with the phylogeneticrelationships between the pandas and the black bears (Fig.2). Analysis of similarity (ANOSIM) supported the PCoA result. The rvalue (ANOSIM, r = 0.93, P < 0.05) betweenthe giant pandas and the red pandas is larger than that (ANOSIM,r = 0.59, P < 0.05) between the giant pandasand the black bears (Table S2).

Principal coordinate analysis of thecommunity structure using Bray-Curtis distances.
Greencircles, blue squares and red triangles represent the gut microbiotas fromthe red pandas, the black bears and the giant pandas, respectively.Distances between symbols on the ordination plot reflect relativedissimilarities in community structures.
These observations were also supported by other measures of distance metrics suchas the ThetaYC11, the weighted Unifrac12 and theMorisita-Horn13 (FigureS1 and S2).
Unique and shared bacterial taxa in the giant and the redpandas
We next sought to examine the shared and unique bacterial taxa between the gutmicrobiotas of the giant and the red pandas using our sequencing data. Thedistributions of the top 10 OTUs of the giant and the red pandas are shown inFig. 3. We used linear discriminant analysis effectsize (LEfSe)14 to identify OTUs differentially representedbetween the red and the giant pandas. While OTU001 and OTU002 were shared by allthe pandas, their relative abundances were differentially represented betweenthe two species of pandas (Fig. 4A). OTU001 is affiliatedwith the genus Streptococcus and significantly higher in the giant pandasthan in the red pandas (Fig. 4B). In contrast, OTU002,classified as Sarcina, a member of the Clostridiaceae family, ismore abundant in the red pandas (Fig. 4C). Both OTUs werealso observed in black bears, but with intermediate relative abundance betweenthe two pandas (Fig. 4B,C). OTU003 was found in 3/5 of thegiant pandas and 4/6 of the red pandas and belonged to the genus ofLactobacillus. OTU10 (Helicobacter) was only observed in thered pandas and was absent from the giant pandas. Whether or not the gutmicrobiotas are involved in the digestion of the highly fibrous diet needsfurther investigation.

Relative abundance of OTUs at the genuslevel in the gut microbiotas from the giant and the redpandas.
Each bar in the stacked bar charts represents therelative abundance of an individual OTU.

OTUs differentially represented betweenthe black bears, the giant and the red pandas identified by linear discriminantanalysis coupled with effect size (LEfSe).
A.Histogram showing operational taxonomic units (OTUs) that are more abundantin the red pandas (blue color), the black bears (red color) or the giantpandas (green color) ranked by linear discriminant analysis (LDA) score. Therelative abundance of OTU001 (more abundant in the giant pandas) and OTU002(more abundant in red pandas) are illustrated in B and C,respectively. The mean and median relative abundance of these OTUs areindicated with solid and dashed lines, respectively.
Discussion
The evolution of the mammal gut microbiotas are affected by several factors. Mueggeet al3, for example, showed that diet has played importantroles in the evolution of the gut microbiota, i.e. the gut microbiotas of thecarnivores are distinct from those of the herbivores and omnivores. Since both thered pandas and the giant pandas have evolved to adapt to the same, highlyspecialized diet (bamboo), it is easy to postulate that they share similar gutmicrobiota. However, our study suggests that despite sharing the same diet, the redpandas and the giant pandas harbor different gut microbiotas. Both the dendrogramand PCoA plot support the divergent evolution of the gut microbiota of these twopandas.
Phylogeny is another factor driving the evolution of the structure of gut microbiotasas reported by several recent studies2,4,15. To put our study intoa phylogenetic context, we incorporated our data with the gut microbiotas of severalother carnivores reported by Muegge et al.3, although notideal due to different DNA extraction methods and regions of the 16S rDNA gene.Nevertheless, we observed similar divergent patterns of the gut microbiotas in thegiant and the red pandas in the combined data set (Figure S3). In a recent study, we reporteddistinct gut microbiotas in the wild and the captive red pandas16.Interestingly, after incorporation of the wild red panda gut microbiota into thisstudy, significant differences in the gut microbiotas between the giant panda andthe wild red pandas were also observed (FigureS4, AMOVA, P < 0.001). Of note, although the divergentevolution of the gut microbiotas in pandas is consistent with their host’sphylogeny, further experiments including more species are required to test thishypothesis. Other unknown host and environmental factors may have also contributedto the divergent evolution of the gut microbiotas in these pandas.
Contrary to a recent study17, which showed diverse bacterialcommunities belonging mainly to Gammaproteobacteria in four giant pandas, weidentified gut microbiotas with much lower diversity and dominated mainly byFirmicutes in the giant and the red pandas. Inconsistency in the gut microbiotas ofpandas was also observed in another study1, which examined the gutmicrobiotas in mammals by sequencing the clone libraries of the 16S gene and showedthat the gut microbiota of the giant panda was dominated by Firmicutes while the gutmicrobiotas of the two red pandas consisted mainly of Gammaproteobacteria. Thediscrepancies between these studies might be attributed to the differences in DNAextraction, hypervariable regions of the 16S rDNA gene, sequencing method and depth,environment and/or other host physiological and genetic factors. Of note, thecommunity diversities of these pandas are low, dominated by one or two OTUs, likelydue to their highly specialized fibrous diet (bamboo) with antibacterialactivities18.
One striking feature of the pandas is their unique bamboo-specialized diet. However,both the giant and the red pandas have short and relatively simple digestive tractand cannot process bamboo efficiently by themselves19, especially thecellulose of the cell walls. Recent culture-independent studies have suggested thepresence of cellulose degraders in both giant20 and red pandas16. It is very possible that although the giant and the red pandaspossess overall different gut microbiota, they do share certain cellulose degradersor degradation pathways that have converged to help with their digestion of bamboo.Future studies using culture based (e.g. cellulose media) or metagenomics (i.e.sequencing the collected gut microbiota instead of just the 16S rRNA gene) basedapproaches are desired to address this question.
In summary, we characterized the gut microbiotas of the red and the giant pandas andfound that, according the 16S rRNA based community structure analysis, the gutmicrobiota of these pandas diverged rather than converged based on the same diet. Wealso identified bacterial taxa deferentially represented between the two species ofpandas. More studies are desired to examine their roles in the hosts’physiology, development, health and disease.
Methods
Ethics Statement
Fecal samples were collected from captive and wild red pandas and black bears inBifengxia Ecological Zoo (Ya’an, Sichuan Province, China). Fecal samplesof the giant pandas were collected in China Conservation and Research Center forthe Giant Panda, Ya’an, Sichuan Province, China. All the samples werecollected by experienced trackers and were immediately frozen in a liquidnitrogen container before transferred to and stored at−80 °C. All animal work was approved by theInstitutional Animal Care and Use Committee of the Sichuan AgriculturalUniversity under permit number DKY-B20130302.
All experiments were performed in accordance with the approved guidelines andregulations.
DNA extraction and pyrosequencing
Frozen fecal samples were thawed on ice and dissected. To avoid soilcontamination, DNA was then extracted from the inner part of the fecal samples(0.25 g) using the MO BIO PowerFecal™ DNA Isolation Kit (MO BIOLaboratories, Carlsbad, CA, USA) according to the manufacturer’sinstructions and the DNA concentration was measured by using Nanodrop (ThermoScientific). DNA pyrosequencing was performed at the Beijing Genomics Institute(BGI Shenzhen, China) via 454 Life Sciences/Roche GS FLX Titanium platform.Briefly, DNA was amplified by using the V1–V3 hypervariable regions ofthe bacterial 16S rRNA gene bar-coded primers (forward: CCGTCAATTCMTTTGAGTTT,reverse: ACTCCTACGGGAGGCAGCAG). The PCR reaction (50 μl)contained 50 ng DNA, 41 μl molecular biology gradewater, 5 μl 10 x FastStart High Fidelity Reaction Buffercontaining 18 mM MgCl2, 1 μl dNTPs(10 mM each), 1 μl Fusion Primer A (10 mM),1 μl Fusion Primer B (10 mM) and 1 μlFastStart High Fidelity Enzyme Blend (5 U/ml). PCR cycles included95 oC for 2 min; 30 cycles of95 oC for 20 s, 50 oCfor 30 s and 72 oC for 5 min; and afinal extension at 72 oC for 10 min.
Sequence analysis
Sequencing reads were processed and analyzed using mothur v1.34 following the 454SOP21 on the mothur wiki webpage ( http://www.mothur.org/wiki/454_SOP) on January 7, 2015. Afterseveral steps of denoising by using the PyroNoise22, Uchime23 and preclustering methods24, high qualitysequences that had a length of at least 200 bp and without sequencingerrors or chimeras were retained and assigned to OTUs using an average neighboralgorithm with a 97% similarity cutoff. OTUs were classified at the genus levelusing the Bayesian method25. The number of reads per sample wasrandomly subsampled to 1,200 to minimize biases caused by sequencing depth.Subsampling to the smallest number of reads was also performed for the two datasets incorporating the wild red pandas and the carnivores from Muegge etal., respectively. For the later, sequences were trimmed to overlap withtheir data during the alignment step.
Good’s coverage and beta diversity measures including Bray-Curtis,Morisita-Horn, Weighted Unifrac and ThetaYC distances were calculated usingmothur. These beta diversity metrics were used to assess the dissimilaritybetween the communities’ structures. Gut microbiota trees were generatedusing the Unweighted Pair Group Method with Arithmetic Mean algorithm based onthe different distance metrics generated by mothur.
Statistical methods
Linear discriminant analysis effect size (LEfSe), which takes into account bothstatistical significance and biological relevance, was conducted to identifyOTUs differentially represented between the red and the giant pandas. AP < 0.05 was considered statistically significant.
Accession numbers
The raw sequences of this study have been deposited in the Sequence Read Archive(accession number SRR1766294). Part of thesequencing data has been published elsewhere to compare the gut microbiota inthe wild and the captive red pandas16.
Additional Information
How to cite this article: Li, Y. et al. The evolution of the gutmicrobiota in the giant and the red pandas. Sci. Rep.5, 10185; doi: 10.1038/srep10185 (2015).
Change history
30 June 2015
The HTML version of this Article previously published omitted Ying Li as a corresponding author. This has now been corrected in the HTML; the PDF version of the paper was correct from the time of publication.
References
Ley, R. E., et al. Evolution of mammals and their gut microbes. Science 320, 1647–1651 (2008).
Ochman, H., et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS biology 8, e1000546 (2010).
Muegge, B. D., et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974 (2011).
Phillips, C. D., et al. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Molecular ecology 21, 2617–2627 (2012).
Godoy-Vitorino, F., etal. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. The ISME journal 6, 531–541 (2012).
Sullam, K. E., et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Molecular ecology 21, 3363–3378 (2012).
Delsuc, F., et al. Convergence of gut microbiomes in myrmecophagous mammals. Molecular ecology 23, 1301–1317 (2014).
Salesa, M. J., Anton, M., Peigne, S. & Morales, J. Evidence of a false thumb in a fossil carnivore clarifies the evolution of pandas. Proceedings of the National Academy of Sciences of the United States of America 103, 379–382 (2006).
Schloss, P. D., et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 75, 7537–7541 (2009).
Bray, J. R. C. & J. T. An ordination of the upland forest communities of southern Wisconsin. Ecological Monographs 27, 325–349 (1957).
Yue, J. C. & Clayton, M. K. A similarity measure based on species proportions. Commun. Stat.-Theor. M 34, 2123–2131 (2005).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology 71, 8228–8235 (2005).
Magurran, A. E. Measuring biological diversity. Oxford: Blackwell PublishingISBN 0-632-05633-9 (2004).
Segata, N., et al. Metagenomic biomarker discovery and explanation. Genome biology 12, R60 (2011).
Moeller, A., et al. Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genome research 23, 1715–1720 (2013).
Kong, F., et al. Characterization of the gut microbiota in the red panda (Ailurus fulgens). PloS one 9, e87885 (2014).
Tun, H. M., et al. Microbial diversity and evidence of novel homoacetogens in the gut of both geriatric and adult giant pandas (Ailuropoda melanoleuca). PloS one 9, e79902 (2014).
Afrin, T., Tsuzuki T., Kanwar R. K. & Wang X. The origin of the antibacterial property of bamboo. Journal of the Textile Institue 103, 844–849 (2012).
Wei, F., Feng, Z., Wang, Z., Zhou, A. & Hu, J. Use of the nutrients in bamboo by the red panda (Ailurus fulgens). Journal of Zoology 248, 535–541 (1999).
Zhu, L., Wu, Q., Dai, J., Zhang, S. & Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proceedings of the National Academy of Sciences of the United States of America 108, 17714–17719 (2011).
Schloss, P. D., Gevers, D. & Westcott, S. L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PloS one 6, e27310 (2011).
Quince, C., Lanzen, A., Davenport, R. J. & Turnbaugh, P. J. Removing noise from pyrosequenced amplicons. BMC bioinformatics 12, 38 (2011).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Huse, S. M., Welch, D. M., Morrison, H. G. & Sogin, M. L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898 (2010).
Cole, J. R., et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–145 (2009).
Acknowledgements
This work was supported by the “1000-Talent Program” in Sichuan andScience Foundation for Youths of Sichuan Province (2013JQ0014) and The NationalNatural Science Foundation of China (31471997) to Y.L. and the Innovative ResearchTeam in University of Sichuan Bureau of Education.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Y.L. and J.Z., Performed the experiments:Y.L., J.Z. and W.G., Contributed reagents/materials/analysis tools: S.H., F.K.,C.W., D.L., H.Z., B.Z., H.X. and M.Y., Wrote the paper: J.Z. and Y.L.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0International License. The images or other third party material in this article areincluded in the article’s Creative Commons license, unless indicatedotherwise in the credit line; if the material is not included under the CreativeCommons license, users will need to obtain permission from the license holder toreproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, Y., Guo, W., Han, S. et al. The evolution of the gut microbiota in the giant and the red pandas. Sci Rep 5, 10185 (2015). https://doi.org/10.1038/srep10185
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10185
This article is cited by
-
Conserve the germs: the gut microbiota and adaptive potential
Conservation Genetics (2019)
-
Conservation metagenomics: a new branch of conservation biology
Science China Life Sciences (2019)
-
The gastrointestinal tract microbiota of northern white-cheeked gibbons (Nomascus leucogenys) varies with age and captive condition
Scientific Reports (2018)
-
Bamboo Specialists from Two Mammalian Orders (Primates, Carnivora) Share a High Number of Low-Abundance Gut Microbes
Microbial Ecology (2018)
-
The Distal Gut Bacterial Community of Some Primates and Carnivora
Current Microbiology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
