
Abstract
Four new ambuic acid derivatives (1–4) and four known derivatives (5–8), were isolated from the solid culture of a plant pathogenic fungus Pestalotiopsis neglecta. Their structures were elucidated by extensive NMR experiments. The absolute configuration of the C-16 secondary alcohol in 1 was deduced via the CD data of the in situ formed [Rh2(OCOCF3)4] complex with the acetonide derivative of 1. The absolute configuration in 3 was assigned by comparison of the experimental and simulated electronic circular dichroism (ECD) spectrum. The NMR data of compound 5 was reported for the first time. In the nitric oxide (NO) inhibition assay, compounds 4, 6 and 7 showed inhibitory activity against the NO production in the lipopolysaccharide (LPS)-induced macrophage with IC50 values of 88.66, 11.20 and 20.80 µM, respectively.
Similar content being viewed by others

Introduction
Nitric oxide (NO) produced by a group of nitric oxide synthases (NOSs) is highly diffusible across cell membranes and modifies many biological molecules1,2. Overproduction of NO is proved to be closely related with many pathogenic diseases, such as inflammation and cancer3. Through reducing NO formation or scavenging NO molecule, NO inhibitors may be used as powerful therapeutic agents4.
The fungi belonging to the genus of Pestalotiopsis have attracted much attention due to their ability in producing diverse secondary metabolites with various biological activities5,6,7,8,9,10. The anticancer drug taxol has been reported from the culture of several species of Pestalotiopsis, which provides an alternative way for the production of this valuable drug11,12. In our searching for natural NO inhibitors from fungi, the pathogenic fungus P. neglecta (FJ–2) was separated from the twig of Camellia sinensis growing in Fujian Province of China. The fungus was grown in a solid-substrate fermentation culture. The EtOAc extract of its solid culture was found to have NO inhibitory activity (IC50 = 250 µg/mL). Chemical investigation on its EtOAc extract afforded four new ambuic acid derivatives (1–4) and four known compounds13,14 5–8. Details of the isolation, structure elucidation and NO inhibitory activity of these compounds are reported herein.
Results and Discussion
The ethyl acetate extract of the solid culture of the fungus P. neglecta was isolated by silica gel column chromatography and ODS column chromatography and finally purified through reversed-phase high performance liquid chromatography (HPLC) to give eight ambuic acid derivatives (1–8). The structures of known compounds (6–8) were determined by NMR data analyses and comparison with the literature data13,14.
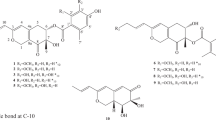
Compound 1 (Figure 1) was isolated as colorless oil. It was assigned the molecular formula C19H26O7 (seven degrees of unsaturation) on the basis of HRESIMS analysis (m/z = 389.1570 [M + Na]+). Its1H and 13C NMR spectra showed resonances for two methyl groups, five methylenes (one oxygenated), three O-methines, six olefinic carbons (three of which were protonated), one oxygenated sp3 quaternary carbon, one carboxylic carbon (δC 171.1, C-1), one α, β-unsaturated ketone carbon (δC 196.0, C-10). These data (Table 1) accounted for all the NMR resonances for 1. The1H-1H COSY NMR data of 1 showed the three isolated spin-systems of C-4–C-3–C-19 (allylic coupling between H-3 with H3-19), C-6–C-7 and C-11–C-17. HMBC correlations from H2-4 to C-5, C-6 and C-10 (δC 196.0), H-6 to C-5, C-7 and C-8 (δC 150.8) and from H2-18 to C-7, C-8 and C-9 (δC 131.9 ), H-11 to C-8, C-9 and C-10 (Figure 2) established the C-5–C-10 cyclohex-2-en-one moiety with the C-4, C-18 hydroxymethyl unit [δH 4.41 d (J = 12.9 Hz), H-18a; δH 4.52 d (J = 12.9 Hz), H-18b; δC 60.3, C-18] and C-11 attached to C-5, C-8 and C-9, respectively. HMBC correlations from H-3 to C-1 (δC 171.1), C-19, from H3-19 to C-1, C-2 and C-3 suggested H3-19 methyl and carboxylic group were both located at C-2. One hydroxy group was located at C-16 [δH 3.73 q (J = 6.2 Hz); δC 68.4, C-16] by correlations from H3-17, H2-14 and H2-15 to C-16. Considering the chemical shifts of C-5 (δC 61.2; 61.3 in ambuic acid13), C-6 (δC 61.1; 61.1 in ambuic acid13) and C-7 (δC 65.9; 66.0 in ambuic acid13) and the unsaturation requirement of 1, the remaining one unsaturation degree was due to an epoxide ring at C-5 and C-6 of the cyclohex-2-en-one. This was furthermore confirmed by the acetonide 1a (Figure 3) that was yielded from the addition of an acetone unit with the hydroxyl groups at C-7 and C-18. On the basis of these data, the planar structure of 1 was proposed.

Chemical structures of compounds 1–8.

Key HMBC  correlations of compounds 1–5.
correlations of compounds 1–5.

CD spectrum of Rh-complex of 1a with the inherent contributions subtracted.
The relative configurations of 1 were deduced by the1H-1H coupling constants and NOESY data. The C-11/C-12, C-2/C-3 double bonds were assigned E-geometry on the basis of the large coupling constant (J = 15.9 Hz) observed between H-11 and H-12 and NOESY correlation of H2-4 with H3-19. The small vicinal coupling constant (J6,7 = 2.8 Hz) suggested a cis orient between H-6 and H-7, the NOESY correlation of H-6 with H2-4 indicated these protons were on the same face of the cyclohex-2-en-one ring. The CD spectrum of 1 showed a positive (350 nm) and a negative (240 nm) Cotton effects, which were similar to those of macrophorin A15, (+)-epoxydon16 and ambuic acid derivatives14, suggesting the 5R, 6R and 7R absolute configuration for 1. The absolute configuration of the C-16 secondary alcohol in 1 was deduced via the CD data of the in situ formed [Rh2(OCOCF3)4] complex with acetonide 1a (Figure 3). The sign of the E band (at ca. 350 nm) can be used to correlate the absolute configuration of a secondary alcohol by applying the bulkiness rule17,18. The Rh complex of 1a displayed a positive Cotton effects at near 350 nm, suggesting the 16S absolute configuration17.
Compound 2 (Figure 1) gave a pseudomolecular ion [M + Na]+ peak at m/z 387.1414 by HRESIMS, consistent with the molecular formula C19H24O7 (eight degrees of unsaturation). Its 1H and 13C NMR spectroscopic data (Table 1)revealed structural similarity to 1, except that one oxygenated methylene [δH 3.73 q (J = 6.2 Hz); δC 68.4, C-16] in 1 was replaced by a ketone (δC 211.8, C-16). It was confirmed by HMBC correlations from H3-17, H2-14 and H2-15 to C-16 (Figure 2). Comparison of the 1H-1H coupling constants and NOESY data of 2 with those of 1 indicated that both compounds possess the same relative configurations at C-5, C-6, C-7 and C-11/C-12, C-2/C-3 double bonds. The absolute configuration of C-5, C-6 and C-7 in 2 was deduced on the basis of CD data. The CD spectrum of 2 (Figure S6) showed a positive (350 nm) and a negative (240 nm) Cotton effects, correlating to the 5R, 6R and 7R configuration.
Compound 3 (Figure 2) was assigned the molecular formula C19H30O7 (five degrees of unsaturation) by HRESIMS (m/z = 393.1887 [M + Na]+). Analysis of its NMR data (Table 1) revealed that 3 possess the similar structural feature to ambuic acid (7), except that the carbonyl group at C-10 in 7 was replaced by an oxygenated methine in 3. Such variation was confirmed by HMBC cross peaks from H-10 to C-4, C-5, C-6, C-8, C-9 and C-11 (Figure 2). The chemical shifts of C-5 (δC 74.2), C-6 (δC 74.0), C-7 (δC 72.5) and C-10 (δC 72.4), the molecular weight of 3, as well as the requirement of unsaturation degree of 3 indicated the opening of the epoxide ring at C-5 and C-6 in 1 and the substitution of hydroxyl group at C-5, C-6, C-7 and C-10. The small vicinal coupling constant (J6,7 = 2.3 Hz) suggested a cis orient between H-6 and H-7, the NOESY correlation of H-6 with H-4b [δH 2.91 dd (J = 15.1, 6.8 Hz)] and H-10 with H-4a [δH 2.70 dd (J = 15.1, 6.8 Hz)] indicated H2-4, H-6, H-7 and H-10 were on the same face of the cyclohex-2-en-one ring.
Since the cyclohex-2-en-one ring system in 3 was relatively rigid, which would significantly affect the CD property, whereas the conformationally flexible side chain had insignificant effect on the CD spectrum of 3, a simplified structure 9 was used for ECD calculations (Figure 4). Considering the relative configuration determined above, one of the two enantiomers (5S, 6R, 7R, 10S)-9a, (5R, 6S, 7S, 10R)-9b should represent the actual absolute configuration of 9. The MMFF94 conformational search followed by B3LYP/6-31G(d) DFT reoptimization afforded four lowest-energy conformers for enantiomers 9a and 9b, respectively (Figure S10). The calculated ECD spectra of enantiomers 9a and 9b were then generated by Boltzmann weighting of the conformers (Figure 4). The absolute configuration of 3 was deduced by comparison of the experimental and simulated electronic circular dichroism (ECD) curves of 9a and 9b. The experimental CD spectrum of 3 was comparable only to the calculated ECD curve of 3a. Therefore, the absolute configuration of 3 was deduced to be 5S, 6R, 7R, 10S.

The experimental CD spectrum of 3 in methanol and the calculated ECD spectra of 9a and 9b.
Structures 9a and 9b represent two possible stereoisomers of 9.
Compound 4 (Figure 1) gave a pseudomolecular ion [M + Na]+ peak at m/z 417.1881 by HRESIMS, consistent with the molecular formula C21H30O7 (seven degrees of unsaturation). Analysis of its NMR data (Table 2) revealed that 4 possess the similar structure to 1, except that C-10 ketone in 1 was reduced to a hydroxyl, the C-16 hydroxyl group was replaced by the hydrogen and the C-18 hydroxy was acetylated. These observations were supported by HMBC cross-peaks from H-10 to C-4, C-5, C-8, C-9, H3-17 to C-15, C-16 and from H2-18, H3-21 to C-20 (Figure 2). The relative configurations for C-5, C-6 and C-7 in 4 were deduced to be the same as those in 1 by comparison of the 1H-1H coupling constants and NOESY data for relevant protons. In 1D NOE experiment of 4, upon irradiation of H-10, enhancements were observed for H2-4 and H-6, suggesting that H-10, H2-4 and H-6 were on the same face of the cyclohex-2-en-one ring. The CD spectrum of 4 (Figure S13) was nearly identical to that of 3, both showing significant negative Cotton effects (CEs) in the regions of 220–260 nm. Therefore, 4 was deduced to have the 5S, 6R, 7R, 10S absolute configuration.
Compound 5 (Figure 1) gave a pseudomolecular ion [M + Na]+ peak at m/z 375.1780 by HRESIMS, consistent with the molecular formula C19H28O6 (six degrees of unsaturation). Analysis of its NMR data (Table 2) revealed that 5 possess the similar structure to 4, except that C-18 O-acetyl group was missed. The relative configurations of 5 were deduced to be the same as those in 4 by comparison of the 1H-1H coupling constants and NOESY data for relevant protons. The CD spectrum of 5 (Figure S16) was matched to those of 3 and 4, both showing significant negative Cotton effects (CEs) in the regions of 220–260 nm. Therefore, compound 5 was deduced to have the 5S, 6R, 7R, 10S absolute configuration. Although previously included in the databases of SciFinder Scholar and PubChem, the information about its isolation and structure assignment was not published. Herein, we report the isolation and NMR data of 5 for the first time.
Ambuic acid (7) and its derivatives have been isolated from the plant endophytic fungus Pestalotiopsis sp. and Monochaetia sp. and from the endolichenic fungus Pestalotiopsis sp. Ambuic acid showed moderate antifungal effects against several plant pathogenic fungi Fusarium solani, Fusarium cubense, Helminthosporium sativum, Diplodia natelensis, Cephalosporium gramineum, Pythium ultimum13 and antimicrobial against the Gram-positive bacterium Staphyococcus aureus14. Ambuic acid also inhibits the biosynthesis of cyclic peptide quormones in Gram-positive bacteria Enterococcus faecalis19. In this study, compounds 2–8 were evaluated for their antifungal activity against other three different pathogenic fungi Aspergillus fumigatus, Aspergillus flavus and Fusarium nivale. They did not show any antifungal activity against tested strains at the concentration of 200 µg/mL.
Macrophages play major roles in inflammation and host defense mechanisms against bacterial and viral infections20. The NO radical produced by the oxidation of L-arginine by NO synthase (NOS) is an effective molecule for the anti-inflammatory and anti-microbial effects of macrophages. However, excessive production of NO may lead to severe injury to host cells and tissues during acute and chronic inflammation. To confirm the bioactive secondary metabolites responsible for the NO inhibitory activity detected in the culture extract of P. neglecta, compounds 1, 4–8 were tested for their inhibitory activity against the NO production in LPS-induced macrophages (Table 3), Compounds 2 and 3 were obtained with fewer amounts (≤1.0 mg) and not enough to test their NO inhibitory activity. In comparison with the positive control of hydrocortisone (IC50 = 53.68 µM), compounds 6 and 7 showed strong NO inhibitory activity with IC50 of 11.20 and 20.80 µM and compound 4 showed weak inhibitory activity with IC50 of 88.66 µM. All other tested compounds exhibited marginal NO inhibitory activity with IC50 larger than 100 µM. It can be presumed that the major secondary metabolites ambuic acid derivative (6) contribute mainly to the NO inhibitory activity detected in the culture. In vivo experiments for 6 are under test. The further studies of anti-inflammatory test for other minor secondary metabolites and more advanced structure-activity relationship of these analogues were not continued due to fewer amounts of these compounds.
In conclusion, eight ambuic acid derivatives including four new secondary metabolites (1–4) were isolated from the culture of the pathogenic fungus P. neglecta. Known compounds 6 and 7 presented strong NO inhibition, which makes them be potential anti-inflammatory agents. The in vivo anti-inflammation assay and action mechanism deserve further investigation.
Methods
General experimental procedures
The optical rotations were measured on a Perkin-Elmer 241 polarimeter (Waltham, USA) and UV spectra were determined on a Thermo Genesys-10S UV-Vis spectrophotometer (Madison, USA). IR data were measured using a Nicolet IS5FT-IR spectrophotometer (Madison, USA). CD spectra were recorded on a JASCO J-815 Spectropolarimeter (Tokyo, Japan). 1H and 13C NMR data were acquired with a Bruker Avance-500 spectrometer (Rheinstetten, Germany) using solvent signals (methanol-d4, δH 3.30/δC 49.9; deuterated chloroform, δH 7.26/δC 77.7) as references. The HMQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. HR-ESI-MS data were acquired using an Agilent Accurate-Mass-Q-TOF LC/MS 6520 instrument (Santa Clara, USA). TLC was carried out on Silica gel HSGF254 and the spots were visualized by spraying with 10% H2SO4 and heating. Silica gel (Qingdao Haiyang Chemical Co., Ltd., Qingdao, People’s Republic of China) and ODS (Lobar, 40–63 µm, Merck, Darmstadt, Germany) were used for column chromatography. Preparative HPLC was performed on an Agilent 1200 HPLC system (Santa Clara, USA) using an ODS column (RP-18, 250 × 10 mm, YMC Pack, 5 μm; detector: UV; Kyoto, Japan) with a flow rate of 2 mL/min. Solvents used for extraction and chromatographic separation were analytical grade.
Fungal material
The pathogenic strain (FJ-2) used in this work was isolated from the twig of Camellia sinensis growing in Fujian Province of China. The fungus was identified on the basis of the DNA sequences of the ITS1-5.8S-ITS2, ITS regions of their ribosomal RNA gene. The sequence data derived from the fungal strain has been submitted and deposited at GenBank with accession number KJ719299. BLAST search result showed that the sequence was similar (99%) to the sequence of Pestalotiopsis neglecta (Thüm.) Steyaert in GenBank with the accession number of JX854541.
The fungal strain was cultured on slants of potato dextrose agar at 25°C for 10 d. Agar plugs were inoculated in 500 mL Erlenmeyer flask containing 120 mL of media (0.4% glucose, 1% malt extract and 0.4% yeast extract, the final pH of the medium was adjusted to 6.5 before sterilization) and incubated at 25°C on a rotary shaker at 180 rpm for 4 d. Large scale fermentation was carried out in 40 bottles of 500 mL Fernbach flasks each containing 80 g of rice and 100 mL of distilled water. Each flask was inoculated with 10.0 mL of culture medium and incubated at 25°C for 40 d.
Extraction and isolation
The fermented rice substrate was extracted thoroughly with ethyl acetate (3 × 5 L) and the organic solvent was concentrated under reduced pressure to afford the crude extract (15.0 g), which was subjected to a silica gel column chromatography (CC) with a gradient of n-hexane-ethyl acetate, dichloromethane-methanol to provide five fractions (A-E).
Fraction C (2.4 g) eluted with dichloromethane-methanol (20:1, v/v) was subjected to ODS CC eluting with a gradient of methanol in water (10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%) to give twenty-one subfractions (C1–C21). Compound 2 (1.0 mg, tR 62.0 min) was purified from subfraction C4 (35 mg) by RP-HPLC using 18% acetontrile in water. Compound 4 (6.9 mg, tR 112.0 min) was obtained from subfraction C10 (87 mg) by RP-HPLC using 30% acetontrile in water. Subfraction C13 (170 mg) was separated by RP-HPLC using 63% methanol in water to afford compound 7 (25.2 mg, tR 37.7 min). Further purification of subfraction C17 (74 mg) on RP-HPLC using 45% acetontrile in water gave compound 6 (4.5 mg, tR 35.0 min).
Fraction D (2.0 g) eluted with dichloromethane-methanol (10:1, v/v) was separated by ODS CC eluted with a gradient of methanol in water (25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%) into twenty-seven subfractions (D1–D27). Further purification of subfraction D5 (50 mg) on RP-HPLC using 30% acetontrile in water gave compound 1 (9.5 mg, tR 6.3 min) and compound 8 (3.5 mg, tR 20.5 min). Compound 3 (0.6 mg, tR 20.8 min) was obtained from subfraction D7 (10 mg) by RP-HPLC using 30% acetontrile in water. Compound 5 (47.5 mg, tR 22.3 min) was purified from subfraction D10 (81 mg) by RP-HPLC using 33% acetontrile in water.
Absolute conguration of the secondary alcohol in 1
In order to determine the absolute configuration of the secondary alcohol in 1, the 7,18-diol in 1 was protected by the derivatization of acetone fork/cross21. Compound 1 (2.0 mg) was treated with 2,2-dimethyoxypropane (1 mL) and pyridinium p-toluene sulfonate (1 mg), then stirred at 30°C for about 8 h under N2 circumstance. The reaction solution was evaporated under reduced pressure and purified by RP-HPLC using 30% acetontrile in water for 8 min following 35% acetontrile in water for 16 min to yield the acetonide 1a (1.0 mg).
A sample of 1a (0.5 mg) was dissolved in a dry solution of the stock [Rh2(OCOCF3)4] complex (1.5 mg) in CH2Cl2 (300 µL) and was subjected to CD measurements. The first CD spectrum was recorded immediately after mixing and its time evolution was monitored until stationary (ca. 10 min after mixing). The inherent CD was subtracted. The observed sign of the E band at around 350 nm in the induced CD spectrum was correlated to the absolute configuration of the C-16 secondary alcohol moiety17,18.
Computational details
Molecular Operating Environment (MOE) ver. 2009.10. (Chemical Computing Group, Canada) software package was used to systematic conformational analyses for compound 9 with the MMFF94 molecular mechanics force field. The MMFF94 conformational analysis was optimized using DFT at B3LYP/6-31G(d) basis set level by Gaussian03 package. The 30 lowest electronic transitions were calculated using the TDDFT methodology at the B3LYP/6-31G(d) level. ECD spectra were stimulated using a Gaussian function with a half-bandwidth of 0.34 eV. The overall ECD spectra were then generated according to Boltzmann weighting of each conformer. The systematic errors in the prediction of the wavelength and excited-state energies are compensated for by employing UV correlation.
Inhibition of NO production by the activated macrophage-like cell line RAW 264.7
Mouse monocyte-macrophages RAW 264.7 (ATCC TIB-71) were purchased from the Chinese Academy of Science. RPMI 1640 medium, penicillin, streptomycin and fetal bovine serum were purchased from Invitrogen (NewYork, USA). Lipopolysaccharide (LPS), DMSO, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and hydrocortisone were obtained from Sigma Chemical Co. (Saint Louis, MO, U.S.A). RAW 264.7 cells were kept in RPMI 1640 medium supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL) and 10% heat-inactivated fetal bovine serum at 37°C in a humidified incubator with 5% CO2 and 95% air. RAW 264.7 cells were passaged by trypsinization until they attained confluence and were used for assays during the exponential growth phase.
Compounds 1, 4–8 were dissolved in DMSO and were further diluted with the culture medium to give a final DMSO concentration of 0.2% in the assay. This concentration of DMSO had no significant effect on the growth of the cell line tested. The procedure of NO inhibition assay was conducted as previously reported. The level of NO was assessed by measuring the accumulation of nitrite (NO2−) using the same method as previously reported22,23. Cytotoxicity was measured using the 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide (MTT) assay method. The inhibition rate was calculated based on the following equation (1).
equation (1):

Antifungal assay
Antifungal assay was conducted in triplicate according to the National Center for Clinical Laboratory Standards (NCCLS) recommendations. The fungi were obtained from China General Microbial Culture Collection (CGMCC) and the American Type Culture Collection (ATCC) and grown on potato dextrose agar (PDA). The fungal were grown on broth cultures that were incubated at 28°C for 48 h and the final suspensions contained 1 × 104 hyphae/mL. Test samples (10 mg/mL at stock solution in DMSO and serial dilutions) were transferred to 96-well plates in triplicate and 200 ul suspensions of fungal were added to each well with alamar blue (10 μL of 10% solution) as indicator. After incubation at 28°C for 48 h, the fluorescence intensity was measured at Ex/Em = 544/590 nm using a microtiter plate reader. The inhibition rate and IC50 were calculated.
compound 1: colorless oil (methanol); [α]25 D 106.9 (c 0.16, methanol); UV (methanol) λmax (log ε): 217 (3.84), 230 (3.89), 273 (3.58) nm; CD (c 9.8 × 10-4 M, methanol): λ (Δε) 219 (5.20), 241 (−0.43), 270 (2.68), 318 (−0.18), 348 (0.11); IR (neat) vmax: 3369, 2965, 2931, 2858, 1682, 1435, 1381, 1264, 1205 cm−1; for 1H NMR and 13C NMR data see Table 1; Positive HR-ESI-MS: m/z 389.1570 [calcd. for C19H26O7Na (M + Na)+, 389.1571].
compound 2: colorless oil (methanol); [α]25 D 111.0 (c 0.1, methanol); UV (methanol) λmax (log ε): 217 (3.85), 230 (3.87), 270 (3.58) nm; CD (c 6.9 × 10-4 M, methanol): λ (Δε) 218 (7.03), 240 (−1.04), 270 (3.41), 316 (−0.19), 350 (0.29); IR (neat) vmax: 3368, 2959, 2923, 2852, 1685, 1438, 1379, 1261, 1207 cm−1; for 1H NMR and 13C NMR data see Table 1; Positive HR-ESI-MS: m/z 387.1414 [calcd. for C19H24O7Na (M + Na)+, 387.1414).
compound 3: colorless oil (methanol); [α]25 D 15.0 (c 0.02, methanol); UV (methanol) λmax (log ε): 230 (3.62) nm; CD (c 2.7 × 10-4 M, methanol): λ (Δε) 215 (1.21), 236 (−11.21), 262 (0.88); IR (neat) vmax: 3350, 2956, 2926, 2854, 1739, 1685, 1412, 1260, 1207 cm−1; for 1H NMR and 13C NMR data see Table 1; Positive HR-ESI-MS: m/z 393.1887 [calcd. for C19H30O7Na (M + Na)+, 393.1884].
compound 4: brown oil (methanol); [α]25 D 3.6 (c 0.23, methanol); UV (methanol) λmax (log ε): 230 (4.41) nm; CD (c 6.2 × 10-4 M, methanol): λ (Δε) 216 (0.17), 236 (−1.80), 259 (0.28), 271 (−0.05), 301 (0.07), 347 (−0.13); IR (neat) vmax: 3391, 2959, 2931, 2859, 1697, 1653, 1434, 1380, 1204 cm−1; for 1H NMR and 13C NMR data see Table 2; Positive HR-ESI-MS: m/z 417.1881 [calcd. for C21H30O7Na (M + Na)+, 417.1884].
compound 5: brown oil (methanol); [α]25 D 20.5 (c 0.22, methanol); UV (methanol) λmax (log ε): 230 (4.21), 283 (3.43) nm; CD (c 1.4 × 10-3 M, methanol): λ (Δε) 213 (1.37), 238 (−1.72), 265 (0.72), 271 (−0.05), 301 (0.07), 347 (−0.13); IR (neat) vmax: 3377, 2957, 2932, 2859, 1691, 1654, 1458, 1420, 1204 cm−1; for 1H NMR and 13C NMR data see Table 2; Positive HR-ESI-MS: m/z 375.1780 [calcd. for C19H28O6Na (M + Na)+, 375.1778].
1a 1H NMR (500MHz, CDCl3): δ 6.74 (1H, t, J = 7.5 Hz, H-3), 5.92 (1H, overlap, H-11), 5.92 (1H, overlap, H-12), 4.79 (1H, br s, H-7), 4.54 (2H, s, H-18), 3.80 (1H, q, J = 6.1 Hz, H-16), 3.67 (1H, d, J = 2.1 Hz, H-6), 2.98 (1H, dd, J = 16.0, 7.5 Hz, H-4b), 2.77 (1H, dd, J = 16.0, 7.5 Hz, H-4a), 2.18 (2H, m, H-13), 1.89 (3H, s, H-19), 1.55 (2H, m, H-14), 1.49 (3H, s, H-21), 1.46 (2H, m, H-15), 1.46 (3H, s, H-22), 1.19 (3H, d, J = 6.1 Hz, H-17). 13C NMR (125MHz, CDCl3): 193.7 (C, C-10), 170.5 (C, C-1), 147.1 (C, C-8), 138.7 (CH, C-12), 136.4 (CH, C-3), 130.6 (C, C-2), 125.5 (C, C-9), 120.2 (CH, C-11), 101.2 (C, C-20), 68.1 (CH, C-16), 65.1 (CH, C-7), 60.9 (CH2, C-18), 59.5 (C, C-5), 55.6 (CH, C-6), 38.8 (CH2, C-15), 33.8 (CH2, C-13), 27.6 (CH2, C-4), 25.4 (CH2, C-14), 24.3 (CH3, C-21), 24.1 (CH3, C-22), 23.7 (CH3, C-17), 12.7 (CH3, C-19). HMBC (500 MHz, CDCl3): H-3 → C-1, C-4, C-5, C-19; H2-4 → C-2, C-3, C-5, C-6; H-6 → C-5, C-7, C-8; H-7 → C-8, C-9; H-11 → C-8, C-9, C-10, C-12, C-13; H-12 → C-9, C-11, C-13; H2-13 → C-11, C-12, C-14, C-15; H2-14 → C-13, C-15, C-16; H2-15 → C-13, C-14, C-16, C-17; H-16 → C-17; H3-17 → C-15, C-16; H2-18 → C-7, C-8, C-9, C-20; H3-19 → C-1, C-2, C-3; H3- 21→ C-20, C-22; H3-22 → C-20, C-21. Positive HR-ESI-MS: m/z 429.1886 [calcd. for C22H30O7Na (M + Na)+, 429.1884].
Additional Information
How to cite this article: Qi,Q.-Y. et al. New ambuic acid derivatives from the solid culture of Pestalotiopsis neglecta and their nitric oxide inhibitory activity. Sci. Rep. 5, 9958; doi: 10.1038/srep09958 (2015).
References
Janakiram, N. B. & Rao, C. V. iNOS-selective inhibitors for cancer prevention: promise and progress. Future Med. Chem. 4, 2193–2204 (2012).
Chen, Y. C., Yang, L. L. & Lee, T. J. F. Oroxylin A inhibition of lipopolysaccharide-induced iNOS and COX-2 gene expression via suppression of nuclear factor-kB activation. Biochem. Pharmacol. 59, 1445–1457 (2000).
Coulter, J. A. et al. Nitric oxide–A novel therapeutic for cancer. Nitric Oxide-Biol. Chem. 19, 192–198 (2008).
Muscara, M. N. & Wallace, J. L. Nitric oxide V. Therapeutic potential of nitric oxide donors and inhibitors. Am. J. Physiol.-Gastroint. Liver Physiol. 276, G1313–G1316 (1999).
Klaiklay, S. et al. Chlorinated chromone and diphenyl ether derivatives from the mangrove-derived fungus Pestalotiopsis sp. PSU-MA69. Tetrahedron 68, 2299–2305 (2012).
Yang, X. L., Awakawa, T., Wakimoto, T. & Abe, I. Induced production of novel prenyldepside and coumarins in endophytic fungi Pestalotiopsis acaciae. Tetrahedron Lett. 54, 5814–5817 (2013).
Harper, J. K. et al. Pestacin: a 1,3-dihydro isobenzofuran from Pestalotiopsis microspora possessing antioxidant and antimycotic activities. Tetrahedron 59, 2471–2476 (2003).
Li, J. et al. Virgatolides A-C, benzannulated spiroketals from the plant endophytic fungus Pestalotiopsis virgatula. Org. Lett. 13, 2670–2673 (2011).
Deyrup, S. T., Swenson, D. C., Gloer, J. B. & Donald, T. W. Caryophyllene sesquiterpenoids from a fungicolous isolate of Pestalotiopsis disseminata. J. Nat. Prod. 69, 608–611 (2006).
Xu, J. et al. Chromones from the endophytic fungus Pestalotiopsis sp. isolated from the Chinese mangrove plant Rhizophora mucronata. J. Nat. Prod. 72, 662–665 (2009).
Strobel, G. et al. Taxol from Pestalotiopsis microspora, an endophytic fungus of Taxus wallachiana. Microbiology-(UK) 142, 435–440 (1996).
Kumaran, R. S., Kim, H. J. & Hur, B. K. Taxol promising fungal endophyte, Pestalotiopsis species isolated from Taxus cuspidata. J. Biosci. Bioeng. 110, 541–546 (2010).
Li, J. Y. et al. Ambuic acid, a highly functionalized cyclohexenone with antifungal activity from Pestalotiopsis spp. and Monochaetia sp. Phytochemistry 56, 463–468 (2001).
Ding, G. et al. Ambuic acid and torreyanic acid derivatives from the endolichenic fungus Pestalotiopsis sp. J. Nat. Prod. 72, 182–186 (2009).
Sassa, T. & Yoshikoshi, H. New terpene-linked cyclohexenone epoxides, macrophorin A, B and C, produced by the fungus caused Macrophoma fruit rot of apple. Agric. Bio. Chem. 47, 187–189 (1983).
Sekiguchi, J. & Gaucher, G. M. Isoepoxydon, a new metabolite of the patulin pathway in Penicillium urticae. Biochem. J. 182, 445–453 (1979).
Frelek, J. & Szczepek, W. J. [Rh2(OCOCF3)4] as an auxiliary chromophore in chiroptical studies on steroidal alcohols. Tetrahedron-Asymmetry 10, 1507–1520 (1999).
Gerards, M. & Snatzke, G. Circular-dichroism, 93. Determination of the absolute-configuration of alcohols, olefins, epoxides and ethers from the CD of their in situ complexes with [Rh2(OCOCF3)4]. Tetrahedron-Asymmetry 1, 221–236 (1990).
Nakayama, J. et al. Ambuic acid inhibits the biosynthesis of cyclic peptide quormones in Gram-positive bacteria. Antimicro. Agents Chemother. 53, 580–586 (2009).
Adams, D. O. & Hamilton, T. A. The cell biology of macrophage activation. Annu. Rev. Immunol. 2, 283–318 (1984).
Zhang, K. et al. Mono- and bis-furanone derivatives from the endolichenic fungus Peziza sp. Fitoterapia 92, 79–84 (2014).
Li, Y. X. et al. A new benzoquinone and a new benzofuran from the edible mushroom Neolentinus lepideus and their inhibitory activity in NO production inhibition assay. Food Chem. 141, 1614–1618 (2013).
Qiu, L. et al. Two new megastigmane glycosides, physanosides A and B, from Physalis alkekengi L. var. franchetii and their effect on NO release in macrophages. Chem. Biodivers. 5, 758–763 (2008).
Acknowledgements
Financial support of the Ministry of Science and Technology of China (2014CB138304 and 2012ZX09301002-003) and Financial support from the National Science Foundation (Grant No. 21472233) and China Ocean Mineral Resources R & D Association (NO. DY125-15-T-07) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
L.B. and H.W.L. designed experiments. Q.Y.Q. and E.W.L. performed the isolation of compounds and analyzed N.M.R. and M.S. data. J.J.H., Y.F.P., K.M., Y.H. and F.Z. conducted experiments. The manuscript was prepared by Q.Y.Q., E.W.L., L.B. and H.W.L. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qi, QY., Li, EW., Han, JJ. et al. New ambuic acid derivatives from the solid culture of Pestalotiopsis neglecta and their nitric oxide inhibitory activity. Sci Rep 5, 9958 (2015). https://doi.org/10.1038/srep09958
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09958
This article is cited by
-
Recent advances in the discovery of bioactive metabolites from Pestalotiopsis
Phytochemistry Reviews (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
