
Abstract
Hepatitis C virus (HCV) infection is the leading cause of chronic liver disease thatcurrently affects at least 170 million people worldwide. Although significantefforts have been focused on discovering inhibitors of a viral polymerase (NS5B) orprotease (NS3), strategies to cure HCV infection have been hampered by the limitedtherapeutic target proteins. Thus, discovery of a novel target remains a majorchallenge. Here, we report a method that combines transcriptome expression analysiswith unbiased proteome reactivity profiling to identify novel host cell responsefactors in HCV infection. A chemical probe for non-directed proteomic profiling wasselected based on genome-wide transcriptome expression analysis after HCV infection,which revealed noticeable alterations related to disulfide bond metabolism. On thebasis of this result, we screened the proteome reactivity using chemical probescontaining thiol-reactive functional groups and discovered a unique labeling profilein HCV-infected cells. A subsequent quantitative chemical proteomic mapping studyled to the identification of a target protein, T-plastin (PLST) and its regulationof HCV replication. Our approach demonstrates both a straightforward strategy forselecting chemical probes to discriminate disease states using a model system andits application for proteome reactivity profiling for novel biomarker discovery.
Similar content being viewed by others


Introduction
Hepatitis C virus (HCV) infection is the leading cause of liver transplantation in theUnited States and almost 80% of patients suffer a persistent chronic infection thatresults in fibrosis, cirrhosis and hepatocellular carcinoma.1 Thecurrently available treatments use a combination of an HCV protease inhibitor withribavirin and PEGylated alpha interferon to disrupt virus replication, but the therapyis effective in only half of the people infected with HCV genotype 1 and even in thosepatients the efficacy is limited.2 Two recently approved drugs targetingthe HCV protease (telaprevir and boceprevir) showed considerably improved curativeeffects,3,4,5 however, there are still unmet needs for moreeffective antivirals. Despite intensive efforts over the last decades, strategies tocure HCV infection have been impeded due to the lack of a detailed understanding of thebiology of the HCV infection process. Most previous attempts were focused on discoveriesof inhibitors of viral polymerases or proteases because of the narrow scope of knowntherapeutic targets.6,7,8 Alternative targets are host cell factorsthat play roles in HCV replication. HCV is a positive-strand RNA virus of theFlaviviridae family that contains 9.6 kb of RNA.9 HCV encodesa single polypeptide protein that is subsequently cleaved into structural (core, E1, andE2) and nonstructural (NS2, NS3, NS4A/B and NS5A/B) subunits by both viral and hostproteases.10 Briefly, viral enzymes (NS2/NS3 and NS3 protease) cleavethe nonstructural proteins from the polypeptide protein to generate mature forms,whereas host cell enzymes are responsible for processing structural proteins.11,12 Thus, host cell factors are closely involved in HCV replication, andthey have high potential as new therapeutic targets for regulating HCV infection.
To examine host cell responses to HCV infection, biologists have utilized conventionalhigh throughput (HTS) techniques, such as gene or proteomic expression profiling.13,14,15,16,17 These approaches have unveiled many important host-HCVinteractions,18,19 but these techniques provide only theperturbations in expression abundance despite the fact that the HCV replication processis highly regulated by various post-translational modifications (PTM) and proteolysis.To directly monitor the catalytic activities of enzymes, an activity-based proteinprofiling (ABPP) method was applied to the protease and fatty acid synthasesuperfamily;20,21 this analysis revealed the differential activityof those enzymes together with several small-molecule regulators.22,23Although ABPP can provide unique insight into the intact metabolic status during HCVinfection, this approach still has drawbacks. First, target enzymes of ABPP probes arelimited to only a few enzyme superclasses at the moment.24,25 Second,the pathological features of many diseases, such as HCV infection, are not wellcharacterized, which makes it difficult to choose proper chemical probes.
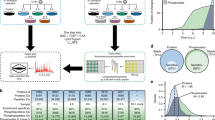
As a complementary method for enzyme activity profiling, undirected proteomic profilinghas unique merits in terms of the diversity of target proteins. It has been reportedthat proteome reactivity can be monitored using various small-moleculeelectrophiles,26,27,28 and the usefulness of identifying functionalcysteine residues29 or discriminating pathogens has beendemonstrated.30 In particular, we found that distinct pathologicalsamples produced fingerprint signatures of proteome reactivity patterns.30 Currently, the major bottleneck step of undirected profiling for disease models isidentification of proper electrophiles to maximize the discriminant signature. Weenvisioned that conventional HTS data could provide insights for selecting desirablechemical probes. Here, we demonstrated a strategy that combines transcriptomeexpression assisted non-directedproteomic profiling (TEAnDPP) to identify host cell response factorsin genotype 2a HCV infection (Figure 1).

Schematic of the transcriptome expression assisted non-directed proteomicprofiling (TEAnDPP) strategy for identifying host cell response factors.
To determine small-molecule electrophiles, we initiated our studies by exploring thetranscriptome analysis of the human hepatoma cell line (Huh7.5) expressing the HCV2asubgenomic replicon (APC140 cells: Huh7.5 cells containing a genotype 2a subgenomicreplicon in bicistronic configuration; HuhHuh7.5/J6/JFHEMCVIRESRlucNeo). The repliconsystem was developed for stable expression of HCV2a proteins in host cells,31 and we chose this system for the ease of culture and for the maintenanceof homogeneity in the viral protein expression. Total RNA was extracted from controlHuh7.5 cells and Huh7.5 cells expressing the HCV2a replicon (APC140 cells) and thegenome-wide transcriptome expression levels were measured using an Illumina Human HT12expression bead array (data are freely available in an NCBI GEO repository: GSE62546).Based on the statistical significance and the fold change values of the expressionlevels, we identified 541 differentially expressed genes (DEGs) out of 47,000 totalgenes with high reproducibility from duplicated experiments (FigS1a-b). Rather than focusing on the strongly responsive genes, we investigatedthe biological functions of all 541 DEGs using DAVID gene enrichment analysis todetermine the general responses of the host cell.32,33 DAVID is abioinformatics tool for integrative functional analysis of a large gene list. Geneontology analysis revealed that biological pathways related to cellular hormonemetabolism and chromatin assembly were considerably perturbed (FigS1c-d). Furthermore, the functional category of the most significantlyenriched DEG cluster was disulfide bond processing (83 genes in 541 DEGs; Table 1). Because gene enrichment analysis showed remarkabledistinctions in cellular thiol metabolism, we hypothesized that thiol-reactive probescould generate differential proteome reactivity signatures upon HCV infection.Therefore, we chose α−iodoacetamide (IA), vinyl sulfone (VS), andbenzyl halide (BH) functional groups that selectively label free thiol groups.
Unlike other diseases that cause dramatic pathological changes, HCV infection tends toinduce subtle and chronic interference in the host cell. In our cell line model, the HCVreplicon expression did not produce noticeable changes in the cell morphology or thetotal proteome band pattern, which was measured using coomassie staining (Fig 2a). To visualize the non-directed proteome reactivity fingerprints inboth cell lines, we used two oppositely charged fluorophores for individual IA, VS, andBH functional groups (Figure S2). We administered each probe at aconcentration of 1 μM for 30 min in live Huh7.5 and APC140 cells,and the cell lysates were separated using SDS-PAGE. Proteome reactivity signatures wereobtained using fluorescence gel imaging with the proper excitation and emission filters(Fig 2b-d). All three electrophiles generated unique proteomereactivity patterns for control Huh7.5 cells (Fig 2b-d: leftlanes). In general, the VS groups exhibited the most intense and numerous bands amongthe three motifs due to their intrinsic high electrophilicity and IA showed faint bandsand the least number of labeled bands. Notably, the undirected protein targets that werelabeled by probes significantly differed depending on the charge state of thefluorophores. Our particular interest was the relative proteome reactivity changesbetween Huh7.5 cells with and without expression of the HCV2a replicon and all 6thiol-reactive probes generated distinct labeling patterns, as we anticipated. Thisobservation was also supporting the finding from the transcriptome analysis that showedthat the reactivities of many cellular thiols were altered by thiol metabolism upon HCVreplication.

Investigation of the non-directed proteome reactivity to iodoacetamide (IA),vinyl sulfone (VS) and benzyl halide (BH) functional groups.
(a) Coomassie staining of Huh7.5 cells without (left) and with expression ofthe HCV2a replicon (right). (b-d) In-gel fluorescence image of the probelabeling in Huh7.5 cells without (left) and with expression of the HCV2areplicon (right).
To investigate the host cell factors that are selectively up-regulated upon HCVreplication, we employed competition-based quantitative chemical proteomic profilinggeared to determine the identity of labeled proteins (Figure S3).Inspired by the competitive isoTOP-ABPP strategy,34 we adapted theprotocol utilizing stable-isotope labeling of amino acids in cell culture (SILAC). SILACinvolves differential labeling of proteins with stable isotopes of different mass togenerate isotopically “heavy” and “light”samples. Because the Flu-IA probe exhibited the most prominent change upon HCV2areplicon expression, we then tried to identify the protein targets of the Flu-IA probe(Fig 2-b). Control Huh7.5 cells were grown in mediumcontaining “heavy” isotopes of arginine (13C6,15N4) and lysine (13C6) and APC140 cells were grown in “light”media. As illustrated in Figure S3, we conducted two-way competitionexperiments in both Huh7.5 cells and APC140 cells. Flu-IA was administered to livecells: either “light” isotope-labeled APC140 cells or“heavy” isotope-labeled Huh7.5 cells at a1 μM concentration for 30 min and whole-cell lysates weresubsequently incubated with an excess amount of biotin polyethylene oxide IA (Biotin-IA,100 μM) to enrich proteins that could form a covalent bond with the IAfunctional group but were not labeled with Flu-IA. Then, cells that were not treatedwith Flu-IA were prepared as a control and the lysates were also labeled with excessBiotin-IA to enrich proteins that could form a covalent bond with the IA functionalgroup, which included Flu-IA targets in this case. The same quantities of proteins weremixed and enriched biotinylated proteins were separated by affinity purification usingavidin-coated agarose beads. Following on-bead trypsin digestion, the peptide mixturesof enriched proteins were separated by nano-flow HPLC and analyzed with using anOrbitrap mass spectrometer.
From the SILAC-based quantitation results, proteins exhibiting competition in both caseswere considered to be non-directed target proteins of Flu-IA. In all, 71 proteins wereidentified from the competition experiment in “light” APC140 cellshaving a SILAC ratio (Heavy/Light) greater than two folds (TableS2a) and 46 proteins were discovered from the competition in“heavy” Huh7.5 cells having a SILAC ratio (H/L) lower than 0.5(Table S2b). Both competition experiments were performed intriplicate and the proteins identified in both cases were 26 proteins with sizesranging from 18.5 kDa to 273.3 kDa (Table S2-c), includingpreviously reported host cell factors for HCV infection, such as chloride channelprotein 1,23 fatty acid synthase,21,35 heat-shock protein90,36 protein disulfide-isomerase,20 and thioredoxinperoxidase.37 From these proteins, we were especially interested inthe one that showed a strong fluorescence band in an SDS gel (Fig2-b). The protein size of the marked band in Figure 3-bwas approximately 70 kDa and there was one protein in that range, plastin-3 (i.e.,T-plastin). We further confirmed the identity of the corresponding band by western blot(Fig S4), but we could not find the exact modification site,possibly due to the low ionization efficiency of the charged modification.

Inhibition of HCV replication.
(a) Effect of T-plastin knock-down measured by Renilla luciferaseactivity. (b) Two target sites of T-plastin RNAi. (c) Dose-dependent HCVreplication inhibition effect of prolonged Flu-IA treatments. (d) Cellviability test by an MTT assay in response to serial concentrations ofFlu-IA treatment. All mean and standard deviation data were obtained fromquadruplicate experiments (N = 4).
The plastin family comprises actin-bundling proteins that are critical to actinregulation in eukaryotes.38 Plastins are evolutionary conserved andexpressed throughout eukaryotes; thus, plastins have been considered one of the keyregulators that have a fundamental cellular function, but functional studies of plastinsare still at an early stage.39,40 Plastins consist of N-terminal EF-handCa2+-binding domains and actin-binding domains (ABD).41 Unlike other ABD-containing proteins, plastins contain two tandem repeats of ABD,which are involved in cross-linking actin filaments into bundles.42
Because HCV NS3 and NS5A proteins interact with microtubules and actin filaments totransfer the replication complex to various subcellular regions,43 wepostulated that up-regulating of T-plastin might have a cooperative influence on HCVreplication. To validate the collaborative effect of T-plastin, we examined thedependence of HCV replication efficiency on perturbations of intact T-plastin. An RNAiknock-down experiment of T-plastin resulted in greater than 50 % inhibition of the HCVreplication efficiency, as indicated by the Renilla luciferase activity encodedin the HCV2a replicon (Fig 3-a). The HCV replication efficiencywas also altered by the Flu-IA modification of T-plastin, which might induceconformational changes similar to those of other endogenous PTMs that disturbed theactin-bundling activity (Fig 3-c).40 In addition,it was previously reported that an actin polymerization inhibitor, cytochalasin D,caused dose-dependent inhibition of the HCV replication efficiency at micromolarconcentrations.44 Taken together, these observations suggested thatthe actin-bundling effect of T-plastin facilitated the HCV replication process, andselective perturbation of T-plastin could be an alternative strategy to treat HCVinfection.
In summary, we have demonstrated a robust strategy that combines transcriptome expressionsignature analysis and non-directed proteome reactivity profiling to discover a novelhost cell response marker for HCV infection. Based on the unique signature of thiolmetabolism, we chose cross-reactive thiol-targeting probes to obtain a proteomereactivity profile and we discovered T-plastin as a novel host cell response factor.Interfering with the expression abundance or exogenous modification of T-plastinattenuates HCV replication, which suggests that modulating this protein may provide astrategy for treating HCV infection. We are currently working on discoveringsmall-molecule ligands that target T-plastin and on applying TEAnDPP to diverseinfectious disease models.
Additional information
Accession codes: Transcriptome expression data are freely available in theNCBI GEO repository (GSE62546). All protein lists and quantitative analysis resultsare also available in the SI.
References
Moradpour, D., Cerny, A., Heim, M. H. & Blum, H. E. Hepatitis C: an update. Swiss Med. Wkly. 131, 291–298 (2001).
Pezacki, J. P., Singaravelu, R. & Lyn, R. K. Host-virus interactions during hepatitis C virus infection: a complex and dynamic molecular biosystem. Mol. Biosyst. 6, 1131–1142 (2010).
Hezode, C. et al. Telaprevir and Peginterferon with or without Ribavirin for Chronic HCV Infection. New Engl. J. Med. 360, 1839–1850 (2009).
Bacon, B. R. et al. Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection. New Engl. J. Med. 364, 1207–1217 (2011).
Jarvis, L. M. The Waiting Game. Chem. Eng. News 88, 12–17 (2010).
Tsantrizos, Y. S. et al. Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection. Angew. Chem. Int. Ed. 42, 1355–1360 (2003).
Gao, M. et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465, 96–100 (2010).
Njoroge, F. G., Chen, K. X., Shih, N. Y. & Piwinski, J. J. Challenges in modern drug discovery: A case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc. Chem. Res. 41, 50–59 (2008).
Choo, Q. L. et al. Isolation of a Cdna Clone Derived from a Blood-Borne Non-a, Non-B Viral-Hepatitis Genome. Science 244, 359–362 (1989).
Penin, F., Dubuisson, J., Rey, F. A., Moradpour, D. & Pawlotsky, J. M. Structural biology of hepatitis C virus. Hepatology 39, 5–19 (2004).
Di Bisceglie, A. M., McHutchinson, J. & Rice, C. M. New therapeutic strategies for hepatitis C. Hepatology. 35, 224–231 (2002).
Moradpour, D., Penin, F. & Rice, C. M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5, 453–463 (2007).
Fang, C. Y. et al. Proteome analysis of human liver carcinoma Huh7 cells harboring hepatitis C virus subgenomic replicon. Proteomics 6, 519–527 (2006).
Su, A. I. et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 99, 15669–15674 (2002).
Hou, J. et al. Gene expression profiling to predict and assess the consequences of therapy-induced virus eradication in chronic hepatitis C virus infection. J. Virol. 88, 12254–12264 (2014).
Chang, K., Wang, T. & Luo, G. Proteomics study of the hepatitis C virus replication complex. Methods Mol. Biol. 510, 185–193 (2009).
Diamond, D. L. et al. Proteomic profiling of human liver biopsies: hepatitis C virus-induced fibrosis and mitochondrial dysfunction. Hepatology 46, 649–657 (2007).
Flajolet, M. et al. A genomic approach of the hepatitis C virus generates a protein interaction map. Gene 242, 369–379 (2000).
Dimitrova, M., Imbert, I., Kieny, M. P. & Schuster, C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 77, 5401–5414 (2003).
Blais, D. R. et al. Activity-Based Proteome Profiling of Hepatoma Cells during Hepatitis C Virus Replication Using Protease Substrate Probes. J. Proteome Res. 9, 912–923 (2010).
Nasheri, N. et al. Modulation of Fatty Acid Synthase Enzyme Activity and Expression during Hepatitis C Virus Replication. Chem. Biol. 20, 570–582 (2013).
Rakic, B. et al. A small-molecule probe for hepatitis C virus replication that blocks protein folding. Chem. Biol. 13, 1051–1060 (2006).
Singaravelu, R., Blais, D. R., McKay, C. S. & Pezacki, J. P. Activity-based protein profiling of the hepatitis C virus replication in Huh-7 hepatoma cells using a non-directed active site probe. Proteome Science 8,5 10.1186/1477-5956-8-5 (2010).
Evans, M. J. & Cravatt, B. F. Mechanism-based profiling of enzyme families. Chem. Rev. 106, 3279–3301 (2006).
Cravatt, B. F., Wright, A. T. & Kozarich, J. W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 77, 383–414 (2008).
Weerapana, E., Simon, G. M. & Cravatt, B. F. Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol. 4, 405–407 (2008).
Shannon, D. A. et al. Investigating the Proteome Reactivity and Selectivity of Aryl Halides. J. Am. Chem. Soc. 136, 3330–3333 (2014).
Adam, G. C., Sorensen, E. J. & Cravatt, B. F. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat. Biotechnol. 20, 805–809 (2002).
Weerapana, E. et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010).
Lee, J. S. et al. Proteome reactivity profiling for the discrimination of pathogenic bacteria. Chem. Commun. 50, 4347–4350 (2014).
Lohmann, V. et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 (1999).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Wang, C., Weerapana, E., Blewett, M. M. & Cravatt, B. F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 11, 79-85 (2014).
Blackham, S. et al. Gene Expression Profiling Indicates the Roles of Host Oxidative Stress, Apoptosis, Lipid Metabolism and Intracellular Transport Genes in the Replication of Hepatitis C Virus. J. Virol. 84, 5404–5414 (2010).
Ujino, S., Yamaguchi, S., Shimotohno, K. & Takaku, H. Heat-shock Protein 90 Is Essential for Stabilization of the Hepatitis C Virus Nonstructural Protein NS3. J. Biol. Chem. 284, 6841–6846 (2009).
Sumida, Y. et al. Serum thioredoxin levels as an indicator of oxidative stress in patients with hepatitis C virus infection. J. Hepatol. 33, 616–622 (2000).
Shinomiya, H. Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines and cancer. Int. J. Cell Biol. 2012, 213492 (2012).
Adams, A. E. M., Shen, W. Y., Lin, C. S., Leavitt, J. & Matsudaira, P. Isoform-Specific Complementation of the Yeast Sac6 Null Mutation by Human Fimbrin. Mol. Cell. Biol. 15, 69–75 (1995).
Morley, S. C. The actin-bundling protein L-plastin: a critical regulator of immune cell function. Int. J. Cell Biol. 2012, 935173 (2012).
Namba, Y., Ito, M., Zu, Y. L., Shigesada, K. & Maruyama, K. Human T-Cell L-Plastin Bundles Actin-Filaments in a Calcium-Dependent Manner. J. Biochem. 112, 503–507 (1992).
de Arruda, M. V., Watson, S., Lin, C. S., Leavitt, J. & Matsudaira, P. Fimbrin is a homologue of the cytoplasmic phosphoprotein plastin and has domains homologous with calmodulin and actin gelation proteins. J. Cell Biol. 111, 1069–1079 (1990).
Lai, C. K., Jeng, K. S., Machida, K. & Lai, M. M. C. Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A. J. Virol. 82, 8838–8848 (2008).
Bost, A. G., Venable, D., Liu, L. & Heinz, B. A. Cytoskeletal requirements for hepatitis C virus (HCV) RNA synthesis in the HCV replicon cell culture system. J. Virol. 77, 4401–4408 (2003).
Acknowledgements
This work was supported by intramural funding from KIST (2Z04070/2E24860-2E25192).The Huh7.5 cell line expressing the HCV2a replicon (APC140 cell line) was kindlyprovided by Dr. Charles Rice and Dr. Takaji Wakita via Apath, LLC.
Author information
Authors and Affiliations
Contributions
J.-S.L. designed the TEAnDPP workflow and conducted the entire proteomic profilingstudy with Y.-H.Y. C.N.Y. and J.-S.L. contributed reagent/material/analysis tools.Y.-H.Y. and J.-S.L. wrote the main manuscript text and J.Y. and Y.-H.Y. preparedfigures 1-3. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info
Supplementary Information
Supplementary Dataset I
Supplementary Information
Supplementary Dataset II
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0International License. The images or other third party material in this article areincluded in the article's Creative Commons license, unless indicatedotherwise in the credit line; if the material is not included under the CreativeCommons license, users will need to obtain permission from the license holder inorder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yoo, YH., Yun, J., Yoon, C. et al. Chemical proteomic identification of T-plastin as a novel host cell response factor inHCV infection. Sci Rep 5, 9773 (2015). https://doi.org/10.1038/srep09773
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09773
This article is cited by
-
Plastin 3 in health and disease: a matter of balance
Cellular and Molecular Life Sciences (2021)
-
Photo-affinity labeling (PAL) in chemical proteomics: a handy tool to investigate protein-protein interactions (PPIs)
Proteome Science (2016)
-
Profiling of Host Cell Response to Successive Canine Parvovirus Infection Based on Kinetic Proteomic Change Identification
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
