
Abstract
Crystal structures of silane have been extensively investigated using ab initio evolutionary simulation methods at high pressures. Two metallic structures with P21/c and C2/m symmetries are found stable above 383 GPa. The superconductivities of metallic phases are fully explored under BCS theory, including the reported C2/c one. Perturbative linear-response calculations for C2/m silane at 610 GPa reveal a high superconducting critical temperature that beyond the order of 102 K.
Similar content being viewed by others

Introduction
Finding high temperature superconductor is one of the important issues in scientific fields. Since N. W. Ashcroft announced the prediction of the metallic hydrogen1, scientists began to explore the hydrogen superconductor. The research about it has never been stopped. Recent theoretical study showed that the Tc of solid metallic hydrogen has achieved 100 K 2. It is still a subject that is full of significant, despite the experimental metallic hydrogen had not found until pressure achieved 300 GPa3,4.
The hydrogen-rich compounds, especially for the hydrides of group IV, are expected high temperature superconductor for comprehending the superconductivity of metallic hydrogen. This attracted more attention on hydrides of group IV. So far, the theoretical studies of GeH4 and SnH4 have predicted high superconductivities with maximal Tc reaching 64 K at 220 GPa for GeH45 and 62 K at 200 GPa for SnH46. In addition, more extensive theoretical and experimental efforts have attempted to reveal the structures and superconductivity of silane. Feng et al.7 found a Pman structure with high superconducting transition temperature Tc 166 K at 202 GPa. Pickard and Needs8 also predicted the structures of SiH4 and studied the structural properties, mentioned the possibility of superconductivity in a C2/c phase. Later Yao et al.9 showed that the Pman structure is in reality not stable by phonon calculations whereas a new C2/c structure is dynamically stable and the superconductivity is close to 50 K at 125 GPa. Eremets et al.10 reported that the metallization of silane occurs between 50 and 65 GPa with the P63 symmetry and the superconducting transition temperature of 17 K at 96 and 120 GPa by using electrical resistance measurements. Chen et al.11 demonstrated that the experimental P63 metallic structure is dynamically unstable by the phonon calculations and a new Cmca phase was predicted. The Cmca silane with a layered network was considered the most likely candidate with the superconducting transition temperature in the range of 20–75 K. Later, Miguel Martinez-Canales et al.12 found the lower enthalpy structures by using the evolutionary algorithm USPEX than those found in previous studies and confirmed that SiH4 is a low-temperature superconductor with a transition temperature of 17 K. These published theoretical and experimental articles indicate that silane is one of the significant materials for comprehending the superconductivity in metallic hydrogen under high pressures. So far, the high-pressure structures of silane are still in discussion and the expected high-temperature superconductivity similar with metallic hydrogen has not been reported. In this paper, the high-pressure crystal structures and potential superconducting property of silane have been extensively explored. Two metallic structures with space groups P21/c and C2/m have been found stable from 383 to 606 GPa and above 606 GPa respectively. Further calculations discover that the superconducting transition temperature has more than 100 K for the C2/m phase. In addition, the superconductivity of C2/c8 has been deeply explored in our work.
Results and Discussion
The crystal structures of silane were predicted with one to six SiH4 formula units per cell. Several competitive candidates for energy with the space groups C2/c (2 molecules/cell), Cmcm (2 molecules/cell), I4/mmm (3 molecules/cell), P21/c (4 molecules/cell), P21/m (4 molecules/cell), P-1 (4 molecules/cell) and C2/m (6 molecules/cell) were obtained. The enthalpies of candidates are plotted as a function of pressures in Figure 1. Above the 300 GPa, there are two competitive enthalpy structures with the C2/c and P21/c symmetry. The maximum enthalpy difference between them is only 0.01 (eV/unit cell) over the pressure range from 300 GPa to 383 GPa, see Figure 1 inset. The C2/c phase predicted in this work is identical with the one proposed by C. J. Pickard and R. J. Needs8, which forms three-dimensional networks. Above 383 GPa, P21/c phase take over C2/c phase and becomes most competitive on enthalpy. As shown in Figure 2(a), Si atoms show a fold layered arrangement with H atoms site around them. There are four SiH4 units with five unequivalent atoms in the conventional cell. One Si atom and four H atoms occupy the crystallographic 4e position with 1 symmetry in this monoclinic crystal. The shortest distance between H atoms is 1.045 Å at 400 GPa, which is longer than the 0.762 Å of the H-H bond length in “H2” unit in the Cmca-12 structure13 of solid hydrogen at the same pressure. P21/c phase keep stable on enthalpy at least to 606 GPa until another competitive phase with C2/m symmetry appears, as shown in Figure 1. C2/m phase obtains monoclinic base-centered lattice and contains six formula units in the conventional cell. There are three unequivalent Si atoms occupy the 4i position with m symmetry, whereas nine H atoms sit on the 8j (site symmetry is 1), 4i (site symmetry is m) and 4g (site symmetry is 2) positions respectively. The shortest distance between two H atoms is 1.009 Å and slightly shorter than 1.045 Å in the P21/c phase. The coordination of Si atoms in C2/c, P21/c and C2/m are all eleven, i.e. each Si atom bond with eleven H atoms, which is quite different with the molecule crystal under low pressure14 and implies some different physical characters. Detail parameters of the structures are listed in Table 1. The decomposition enthalpies reference to Cmca-1213 structure of H2 (below 500 GPa), I41/amd13,15 structure of H2 (500–1000 GPa), Fm-3m16 structure of Si and C2/c17 structure of SiH3 can also be seen in Figure 1 (The comparison with SiH4(H2)2 are shown in supplementary information). It is noteworthy that the enthalpies of the three structures are lower. Moreover, P21/c and C2/m structures are found to be energetically much superior to previous structures7,8,9,11,12. Therefore, three monoclinic (C2/c, P21/c and C2/m) phases can be taken as energetically stable structures of silane under high pressure range, see Figure 1.

Enthalpy difference curves of Silane.
Calculated enthalpies per SiH4 unit of various structures relative to our predicted Cmcm structure as a function of pressure range from 300–1000 GPa. Inset: Enthalpies in the pressure range from 310 GPa to 430 GPa.

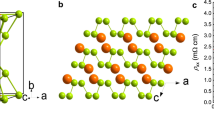
Structures of silane.
Big atoms depict Si, while small atoms represent H. (a) Conventional cell of P21/c silane, stable between 383 and 606 GPa. (b) Primitive cell of C2/m silane, favored above 606 GPa.
The mechanical stability of structure can provide insight into the stability of materials. To evaluate the mechanical stability of the C2/c, P21/c and C2/m phases, elastic constants have been calculated and listed in Table 2. According to the mechanical stability criteria, the crystal deformation energy is positive, this means the determinants of elastic constants matrix Cij should be positive18. Considering the crystal symmetry, the mechanical stability of expression will be further simplified19. It can be found that the elastic constants of these three structures satisfy the mechanical stability criteria, indicating that these three structures are mechanically stable. The phonon band structure and projected phonon density of states (PHDOS) of the three phases at selected pressures are presented in Figure 3. Absence of any imaginary frequency in the Brillouin zone establishes the dynamical stability. The PHDOS of these three structures shows that the heavier Si atoms dominate the low-frequency vibrations and the lighter H atoms contribute significantly to the high-frequency modes.

The phonon band structure and projected phonon DOS charts.
(a) C2/c phase at 300 GPa. (b) P21/c phase at 400 GPa. (c) C2/m phase at 610 GPa.
To analysis the electronic properties of C2/c, P21/c and C2/m phases, we first calculated the electronic density of states (DOS). As shown in Figure 4, they are all metals with large total DOS at Fermi level. Specially, for C2/m structure, the total DOS of Fermi level is significantly higher than the other structures. These high DOS values might favor the superconducting behavior. From the Figure 4(a–c), H atoms contribute more to DOS than the Si atoms below Fermi level and contribute less to DOS than the Si atoms above Fermi level. With increasing pressure, the contributions from atoms H and Si do not significantly change. The electron localization functions (ELF)20 of the C2/c, P21/c and C2/m phases are calculated at 300 GPa, 400 GPa and 610 GPa, respectively. The isosurface plots at ELF = 0.5 are shown in Figure 4(d–f). The electron-gas-like distribution in this space are connected, which conforms the three structures are metallic.

The electronic projected DOS and electron localization function isosurface maps.
Electronic projected DOS of (a) C2/c phase at 300 GPa, (b) P21/c phase at 400 GPa and (c) C2/m phase at 610 GPa, respectively. Electron localization function isosurface maps for (d) C2/c structure at 300 GPa, (e) P21/c structure at 400 GPa, (f) C2/m structure at 610 GPa. The yellow isosurface represents an ELF value of 0.5. Blue and orange spheres represent Si and H, respectively.
The electron-phonon coupling strength (EPC) λ and the logarithmic average phonon frequency ωlog of the three structures were calculated to explore the possible superconductivity of SiH4. The Eliashberg phonon spectral function α2F(ω) and the λ as a function of frequency are shown in Figure 5. The λ of C2/c structure at 300 GPa, P21/c structure at 400 GPa and C2/m structure at 610 GPa are 0.69, 0.66 and 1.18, respectively. The superconducting critical temperature can be estimated from the Allen–Dynes modified McMillan equation21

which has been found to be highly accurate for many materials with λ < 1.5. The Coulomb pseudopotential μ* is taken as 0.13 for hydrogen dominant metallic alloys by Ashcroft22, resulting that the estimated Tc of C2/c, P21/c and C2/m at 300 GPa, 400 GPa and 610 GPa are 29.65 K, 31.57 K and 106.31 K, respectively. Subsequently, the contributions to EPC λ of each atom are analyzed. As for C2/c, P21/c and C2/m, Si vibrations provide a contribution of 36%, 34% and 37% respectively, while the H translational vibrations contribute for nearly 64%, 66% and 63% respectively. The result shows that the element H plays a significant role in the EPC λ.

The Eliashberg phonon spectral function α2F(ω), the electron-phonon integral λ and the three-dimensional Fermi surface of C2/c, P21/c and C2/m at 300, 400 and 610 GPa, respectively.
The trend of Tc with pressures for these three structures was explored for further investigation. Tc, ωlog and λ along with the changes of pressures are displayed in Figure 6. With increasing pressure, the decrease of λ plays an important role to the downward trend of Tc. Similar phenomena can be observed in other hydrogen-rich materials10,23,24,25. We then use the rigid-muffin-tin (RMT) theory of Gaspari and Gyorffy26 for further analysis this variation. MaMillan's strong coupling theory22,27 defines an electron-phonon coupling constant by

where M is the atomic mass. The DOS at the Fermi level N(ϵF), the square of the electron-ion matrix element 〈I2〉 and the average phonon frequency  of the three phases at several pressures are shown in Figure 6. From the Figure,
of the three phases at several pressures are shown in Figure 6. From the Figure,  increases generally with pressure, meanwhile N(ϵF) decrease (the values between C2/m at 610 GPa and 700 GPa are closely identical). The tendency of the two parameters made λ become lower and lower, which lead to the decrease of Tc with increasing pressure.
increases generally with pressure, meanwhile N(ϵF) decrease (the values between C2/m at 610 GPa and 700 GPa are closely identical). The tendency of the two parameters made λ become lower and lower, which lead to the decrease of Tc with increasing pressure.

Calculated Tc (K), the logarithmic average phonon frequency ωlog (K), electron-phonon coupling strength λ, the DOS at the Fermi level N(ϵF) (state/eV/Cell), the square of the electron-ion matrix element 〈I2〉 (eV/Å)2 and the average phonon frequency  (THz) of the three phases as a function of pressure.
(THz) of the three phases as a function of pressure.
The red line respect C2/c phase at 300–380 GPa. The blue line respect P21/c phase at 400–600 GPa. The green line respect C2/m phase at 610–800 GPa.
We also realized that Tc magnitude of C2/m is beyond 102 K which is higher than other two phases that promoted us to explore the underlying superconducting mechanism. From Figure 6, the trend of λ along with pressures is consistent with Tc. So, λ played an important role in the superconducting critical temperature. The calculated EPC λ of C2/m phase at 610 GPa is 1.18, which is much higher than the values of 0.69 and 0.66 of two phases. The bigger EPC λ can directly contribute to higher Tc. Furthermore, formula (2) defined by MaMillan's strong coupling theory can be used to analyze the mechanisms. Comparing the contribution to λ in the phase of C2/m and C2/c, Figure 6 reveals that N(ϵF) of C2/m contributes about four times than the one of C2/c, meanwhile the two factors of  and 〈I2〉 together contribute about half of C2/c. Although there are two disadvantages of parameters in C2/m according formula (2), the higher N(ϵF) sustains the higher λ as a whole. As for C2/m and P21/c, there are no obvious differences on the values of
and 〈I2〉 together contribute about half of C2/c. Although there are two disadvantages of parameters in C2/m according formula (2), the higher N(ϵF) sustains the higher λ as a whole. As for C2/m and P21/c, there are no obvious differences on the values of  and 〈I2〉 and λ can be dominated by N(ϵF) as well. As shown in Figure 6, N(ϵF) in C2/m is much higher than the ones in other two structures. Based on an overall analysis of contributions from parameters in two formulas including the contribution from ωlog, N(ϵF) play an important role to the much higher Tc than other phases.
and 〈I2〉 and λ can be dominated by N(ϵF) as well. As shown in Figure 6, N(ϵF) in C2/m is much higher than the ones in other two structures. Based on an overall analysis of contributions from parameters in two formulas including the contribution from ωlog, N(ϵF) play an important role to the much higher Tc than other phases.
It is consistent with the calculated three-dimensional Fermi surface which is shown in Figure 5. There are four electronic bands across the Fermi surface for all the three phases and identified by different colors. As shown in Figure 5, the C2/m phase is filling with much more Fermi surfaces in the brillouin zone than the two other phases, which is corresponding with the high level of N(ϵF). Therefore, the Fermi surface also support the conclusion that the high values of N(ϵF) is significant to sustain high Tc.
Conclusion
In summary, we have extensively investigated crystal structures and superconductivity of silane. The P21/c and C2/m structures are found which are thermodynamically, mechanically and dynamically stable. The superconducting critical temperature Tc of the C2/c phase at 300 GPa and P21/c at 400 GPa are 29.65 K and 31.57 K. The superconductivity of the C2/m phase with a transition temperature 106.31 K is found to be mainly attributed to the strong electron-phonon coupling due to the high electronic density of states at the Fermi level.
Methods
The most stable structures of silane under high pressures were performed using evolutionary algorithm, as implemented in the USPEX code28. This approach has been successfully used in the study of many materials at high pressures29,30. The structural relaxations have been performed within the framework of the generalized gradient approximation (GGA)31 with the Perdew–Burke–Ernzerhof parameterization for the exchange-correlation functional to DFT by using the projector augmented-wave(PAW) method32, as implemented in ab initio VASP code33. The structures were relaxed at a high cutoff energy of 1000 eV and a Brillouin zone sampling grid of spacing 2π × 0.03 Å−1. The lattice dynamics and electron-phonon coupling have been computed with QUANTUM-ESPRESSO34. We used Vanderbilt-type ultrasoft pseudopotentials35 with a cutoff energy of 40 Ry. Phonon frequencies were calculated based on the density functional linear-response method36. A Monkhorst-Pack(MP)37 Brillouin zone sampling grid of spacing 2π × 0.025 Å−1 with Gaussian smearing of 0.02 Ry were used for the phonon calculations, at 4 × 4 × 4, 5 × 4 × 3 and 3 × 3 × 2 q-point mesh for C2/c, P21/c and C2/m for the electron-phonon interaction matrix element, respectively. All the convergences of the plane-wave basis set and MP sampling are carefully examined by employing higher kinetic energy cutoffs and denser grids sets. The tests of cutoff energy and the validity of potential functions are shown in supplementary information.
References
Ashcroft, N. W. Metallic hydrogen: a high-temperature superconductor? Phys. Rev. Lett. 21, 1748 (1968).
Cudazzo, P. et al. Ab initio description of high-temperature superconductivity in dense molecular hydrogen. Phys. Rev. Lett. 100, 257001 (2008).
Loubeyre, P., Occelli, F. & LeToullec, R. Optical studies of solid hydrogen to 320 GPa and evidence for black hydrogen. Nature 416, 613–617 (2002).
Goncharov, A. F., Gregoryanz, E., Hemley, R. J. & Mao, H. Spectroscopic studies of the vibrational and electronic properties of solid hydrogen to 285 GPa. Proc. Natl. Acad. Sci. U.S.A. 98, 14234–14237 (2001).
Gao, G. et al. Superconducting high pressure phase of germane. Phys. Rev. Lett. 101, 107002 (2008).
Gao, G. et al. High-pressure crystal structures and superconductivity of stannane (SnH4). Proc. Natl. Acad. Sci. U.S.A. 107, 1317–1320 (2010).
Feng, J. et al. Structures and potential superconductivity in SiH4 at high pressure: en route to “metallic hydrogen”. Phys. Rev. Lett. 96, 017006 (2006).
Pickard, C. J. & Needs, R. J. High-pressure phases of silane. Phys. Rev. Lett. 97, 045504 (2006).
Yao, Y., Tse, J. S., Ma, Y. & Tanaka, K. Superconductivity in high-pressure SiH4 . Euro. Phys. Lett. 78, 37003 (2007).
Eremets, M. I., Trojan, I. A., Medvedev, S. A., Tse, J. S. & Yao, Y. Superconductivity in hydrogen dominant materials: silane. Science 319, 1506–1509 (2008).
Chen, X. et al. Superconducting behavior in compressed solid SiH4 with a layered structure. Phys. Rev. Lett. 101, 077002 (2008).
Martinez-Canales, M. et al. Novel structures and superconductivity of silane under pressure. Phys. Rev. Lett. 102, 087005 (2009).
Pickard, C. J. & Needs, R. J. Structure of phase III of solid hydrogen. Nat. Phys. 3, 473–476 (2007).
Degtyareva, O. et al. Crystal structure of SiH4 at high pressure. Phys. Rev. B 76, 064123 (2007).
Johnson, K. A. & Ashcroft, N. W. Structure and bandgap closure in dense hydrogen. Nature 403, 632–634 (2000).
Katzke, H., Bismayer, U. & Tolédano, P. Theory of the high-pressure structural phase transitions in Si, Ge, Sn and Pb. Phys. Rev. B 73, 134105 (2006).
Jin, X. et al. Superconducting high-pressure phases of disilane. Proc. Natl. Acad. Sci. U.S.A. 107, 9969–9973 (2010).
Nye, N. F. Physical Properties Of Crystal. 142–143 (Oxford Univ. Press, Oxford, 1985).
Wu, Z. et al. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 76, 054115 (2007).
Becke, A. D. & Edgecombe, K. E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 92, 5397–5403 (1990).
Allen, P. B. & Dynes, R. C. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 12, 905–922 (1975).
Ashcroft, N. W. Hydrogen Dominant metallic alloys: high temperature superconductors? Phys. Rev. Lett. 92, 187002 (2004).
Flores-Livas, J. A. et al. High-Pressure Structure of Disilane and Their Superconducting Properties. Phys. Rev. Lett. 108, 117004 (2012).
Kim, D. K. et al. Predicted Formation of Superconducting Platinum-Hydride Crystals under Pressure in the Presence of Molecular Hydrogen. Phys. Rev. Lett. 107, 117002 (2011).
Zhou, X. F. et al. Superconducting high-pressure phase of platinum hydride from first principle. Phys. Rev. B 84, 054543 (2011).
Gaspari, G. D. & Gyorffy, B. L. Electron-Phonon Interactions, d Resonances and Superconductivity in Transition Metals. Phys. Rev. Lett. 28, 801–805 (1972).
McMillan, W. L. Transition Temperature of Strong-Coupled Superconductors. Phys. Rev. 167, 331–344 (1968).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, 244704 (2006).
Zhu, Q. et al. Stability of xenon oxides at high pressure. Nat. Chem. 5, 61–65 (2013).
Zhang, W. et al. Unexpected stable stoichiometries of sodium chlorides. Science 342, 1502 (2013).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made sample. Phys. Rev. Lett. 77, 3865–3858 (1996).
Kresse, G. & Joubert, D. From ultrasoftpseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. : Condens. Matter 21, 395502 (2009).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized formalism. Phys. Rev. B 41, 7892–7895 (1990).
Baroni, S., Giannozzi, P. & Testa, A. Green's-function approach to linear response in solids. Phys. Rev. Lett. 58, 1861–1864 (1987).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2011CB808200), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1132), the National Natural Science Foundation of China (Nos. 51032001, 11074090, 10979001, 51025206, 11174102) and National Found for Fostering Talents of basic Science (No. J1103202). Parts of calculations are performed at the High Performance Computing Center (HPCC) of Jilin University.
Author information
Authors and Affiliations
Contributions
T.C. initiated the project. H.Z. performed the first principle calculations and prepared all figures. H.Z., X.J. and T.C. analyzed the data and wrote the manuscript text. Y.Z.L., Q.Z., Y.X.L., Q.L., K.B., D.L. and B.L. reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, H., Jin, X., Lv, Y. et al. High-temperature Superconductivity in compressed Solid Silane. Sci Rep 5, 8845 (2015). https://doi.org/10.1038/srep08845
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08845
This article is cited by
-
Strong correlation between electronic bonding network and critical temperature in hydrogen-based superconductors
Nature Communications (2021)
-
Structural, mechanical and electronic properties and hardness of ionic vanadium dihydrides under pressure from first-principles computations
Scientific Reports (2020)
-
Pressure dependence of electronic structure and superconductivity of the MnX (X = N, P, As, Sb)
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
