
Abstract
The Three Gorges Dam has significantly altered ecological and environmental conditions within the reservoir region, but how these changes affect bacterioplankton structure and function is unknown. Here, three widely accepted metagenomic tools were employed to study the impact of damming on the bacterioplankton community in the Xiangxi River. Our results indicated that bacterioplankton communities were both taxonomically and functionally different between backwater and riverine sites, which represent communities with and without direct dam effects, respectively. There were many more nitrogen cycling Betaproteobacteria (e.g., Limnohabitans) and a higher abundance of functional genes and KEGG orthology (KO) groups involved in nitrogen cycling in the riverine sites, suggesting a higher level of bacterial activity involved in generating more nitrogenous nutrients for the growth of phytoplankton. Additionally, the KO categories involved in carbon and sulfur metabolism, as well as most of the detected functional genes also showed clear backwater and riverine patterns. As expected, these diversity patterns all significantly correlated with environmental characteristics, confirming that the bacterioplankton communities in the Xiangxi River were really affected by environmental changes from the Three Gorges Dam. This study provides a first comparative metagenomic insight for evaluating the impacts of the large dam on microbial function.
Similar content being viewed by others

Introduction
As one of the most abundant types of organisms on Earth, bacterioplankton have been found almost anywhere water exists1. They are an extremely diverse and highly dynamic group whose activities directly influence the water conditions in their immediate environment as well as having profound effects on biospheric biogeochemical cycling2. In turn, bacterioplankton communities also undergo significant shifts in composition or abundance at both spatial and temporal scales in response to environmental variations3,4,5. Construction of river dams can significantly alter river hydrology and aquatic ecology upstream and downstream of the dam6, creating environments that have been significantly altered physically, chemically and biologically. These changes can, in turn, greatly affect the aquatic microorganisms living in the reservoir systems, especially environmentally sensitive organisms, such as bacterioplankton7,8,9. However, most of the investigations on the effects of dams to planktonic organisms has focused on community composition and has primarily relied on morphological methods for examining community changes. Fortunately, the recently developed culture-independent rRNA gene sequencing has already offered a useful glimpse into microbial diversity10,11,12,13,14 and the more powerful high-throughput metagenomic sequencing technology is a further key innovation for exploring community composition and function in microbial ecology15. However, knowledge regarding microbial function in response to dam impacts is still lacking.
We know that similar communities may have strikingly divergent functions and distinct community configurations can perform similar functions16. Consequently, an understanding of the community composition is insufficient to reveal the functional significance of a community in a particular environment. Recently, the whole metagenomic shotgun sequencing and high-throughput GeoChip functional gene microarray analysis have become attractive approaches to examine the functional properties of microbial communities17,18,19. For aquatic environments, most attention has focused on ocean environments14,20 and a few freshwater lakes15,21, but very little attention has been paid to bacterioplankton metagenomes in river/reservoir systems22. The Xiangxi River is an ideal river/reservoir system to address the impacts of dams on bacterioplankton since it is the nearest large tributary to the Three Gorges Dam. It has clear backwater and riverine areas, which represent bacterioplankton communities with or without direct dam effects, respectively.
Since the impoundment of the Three Gorges Reservoir began in June 2003, the backwater area of the Xiangxi River (the region with water being held or pushed back by the dam) has been transformed from a riverine system into a lacustrine system, accompanied by significant ecological and environmental changes. For example, the water level has increased more than 40 m; the water flow velocity has dropped significantly23; and water blooms have been detected in spring at some newly formed bays24,25. Although phytoplankton succession and the process underlying blooms in the Xiangxi River have been frequently studied24,25,26, our understanding of damming effects on planktonic microorganisms is largely restricted to phytoplankton shifts and related environmental changes, while changes in bacterioplankton have been largely ignored. A few studies have addressed the bacterial community composition along the Yangtze River using more course-level culture-independent methods such as PCR-DGGE and clone libraries8,27,28; but high-throughput sequencing and functional profile-based approaches have not been involved. As such, there are still questions regarding the structural and functional responses of bacterioplankton to environmental changes caused by the Three Gorge Dam and the biogeochemical roles they play in this newly formed reservoir system.
To gain insight into the bacterioplankton community structure and the functional potential of this community in the Xiangxi River after the damming and to better understand bacterioplankton roles in biogeochemical cycling, we performed 16S rRNA gene sequencing, functional gene array (GeoChip 5.0) analysis and metagenomic shotgun sequencing on six bacterioplankton communities from different areas of the Xiangxi River. Specifically, we addressed the following questions in this study: (i) are the bacterioplankton communities in backwater and riverine sites taxonomically and/or functionally different due to effects from the dam; (ii) what is the relationship between the bacterioplankton community structure (both phylogenetic and functional) and the environmental conditions changed by the damming regulation; and (iii) how is the community functional potential altered by the dam, specifically those functions involved in the cycling of key natural elements (e.g., carbon, nitrogen and sulfur)?
Results
Environmental characteristics, phyto- and zooplankton composition
Physicochemical analysis indicated that the nutrient concentrations of total phosphorus (TP), phosphate phosphorus (P-PO4) and chemical oxygen demand (COD) were significantly lower in the backwater sites than in the riverine sites (t-test, P < 0.05). However, the concentrations of total nitrogen (TN), ammonium nitrogen (N-NH4) and nitrate nitrogen (N-NO3) were significantly higher in the backwater area (Table S1). These nutrient variations, which have been shown to be significantly associated with bacterioplankton28,29, could be directly or indirectly affected by the Three Gorges Dam. However, some other factors such as pH, turbidity, transparency, temperature, dissolved oxygen and conductivity showed no statistical difference between the two investigated areas (Table S1) as showed in Figure 1.

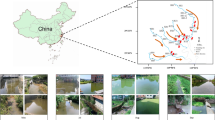
Location of the six sampling sites along the Xiangxi River in the Three Gorges Reservoir (created using Adobe Photoshop 8.01 software).
The estuary, midstream and upstream sites along the Xiangxi River are abbreviated as XXR_E, XXR_M and XXR_U, respectively; the other three sites are the Wujia Bay (WJB), the estuary of the Baisha River (BSR_E) and the estuary of the Shendu River (SDR_E).
Based on morphological identification (generally to the genus or species level), Cyclotella sp. were the most abundant algae (108–109 ind. L−1) in the Xiangxi River (Table S2). Although only Pyrrophyta and Pandorina spp. abundances showed significant difference between backwater and riverine sites (t-test, P < 0.05), the algal diversity in riverine sites was much higher than in the backwater sites (Table S2). There were many more zooplankton taxa (especially protozoa and rotifer) in the riverine sites (Table S2). Furthermore, the riverine sites generally harbored a significantly higher abundance of rotifer species (e.g., Trichocerca pusilla, Polyarthra vulgaris, Synchaeta stylata, Anuraeopsis fissa) and Ciliophora. However, there were more large zooplanktonic cladocerans (e.g., Daphnia hyaline, Bosmina longirostris) and copepods (e.g., Sinocalanus dorrii) in the backwater sites than in the riverine sites (Table S2).
Bacterioplankton composition determined using high-throughput sequencing
The valid sequences obtained could be assigned to 1,368 operational taxonomic units (OTUs) using the UClust method. The overlap of detected OTUs between backwater and riverine sites (33.71% ± 1.55%) was much lower than that observed within the backwater or riverine sites (54.98% ± 3.83%, one-way ANOSIM, P = 0.0006). These OTUs clustered into 22 phyla, 51.17% of which were classified as Proteobacteria (Fig. S1), followed by the Bacteroidetes (22.22%) and Actinobacteria (15.28%). Among all the 18 genera that each accounted for more than 1% of the total bacterial abundance, 8 showed significant differences between the backwater and riverine sites (mainly from Bacteroidetes and Proteobacteria, Table S3.1). Among all dominant OTUs (each > 1% of the total bacterial abundance), more than a half had abundances that were significantly different between the backwater and riverine sites (mainly from Proteobacteria and Actinobacteria, Table S3.2). Rarefaction analysis indicated that sequencing for all of the communities investigated here reached near saturation at a genetic distance of 3% (Fig. S2).
The Chao value, Shannon, inverse Simpson richness, Pielou and Simpson evenness all indicated that the α-diversity of bacterioplankton was significantly higher (t-test, P < 0.01) in the backwater sites than in the riverine sites (data not shown). The β-diversity also suggested that bacterioplankton communities within the backwater/riverine sites were much more similar than between the backwater and riverine sites. Although the Bray-Curtis distance based dissimilarity tests already confirmed that the community differences between the two investigated areas were significant (PERMANOVA, F = 0.755, P = 0.001). We further applied the Raup-Crick probability-based index (SRC)30 to confirm whether differences among the communities were significant (SRC < 0.05), or due to chance (0.05 < SRC < 0.95). The SRC provides a measure of statistically significant similarity and dissimilarity at the 95% confidence level and therefore is less affected by sampling bias31. An additional feature of this index is that biogeographic data are weighted on the basis of the frequency of occurrence and widespread taxa do not disproportionately influence the similarity32. We found that all SRC values between the backwater and riverine sites were less than 0.05, confirming that the backwater communities were significantly different from those in the riverine sites. But all SRC values within the backwater or riverine sites were larger than 0.95 (significantly similar).
In terms of relative abundance, Proteobacteria (mainly Betaproteobacteria) was the most abundant bacterial phylum in the Xiangxi River (accounting for 32.93%–74.67% among different sites) and had a significantly higher abundance in the riverine sites than in the backwater sites (Table S4). Actinobacteria was significantly higher in the backwater sites (18.78%–34.75%) than in the riverine sites (Table S4). The canonical correspondence analysis (CCA) indicated that the N-NH4, N-NO3, COD and temperature showed significant relationships with taxonomic composition (Fig. 2A). The first two axes explained 71.4% of the taxonomic information. Variance partitioning CCA further suggested that nitrogen sources of N-NH4 and N-NO3 may be the most important factors affecting taxonomic composition (Fig. 3A).

Canonical correspondence analysis (CCA) shows the relationships between environmental variables and the bacterial OTUs (A) or functional genes (B).
Only variables that were significantly correlated with the community (forward selection with Monte Carlo test, P < 0.05) are shown. Abbreviations: TP, total phosphorus; N-NH4, ammonium nitrogen; N-NO3, nitrate nitrogen; COD, chemical oxygen demand. The full name of each sampling site is shown in Figure 1.

Variance partitioning canonical correspondence analysis (CCA) shows the relative effects of multiple variables on the composition of bacterial taxa (A) and functional genes (B).
The squares represent the effect of individual variables by partitioning out the effects of the other variables. The ellipses between the squares represent the combined effects from the variables on either side of the ellipse. The combined effects of all variables are shown by the ellipse in the center. The square at the bottom of each figure represents the effect that could not be explained by any of the variables tested. Abbreviations: TP, total phosphorus; N-NH4, ammonium nitrogen; N-NO3, nitrate nitrogen; COD, chemical oxygen demand.
Functional gene diversity detected using GeoChip 5.0
Microarrays detected significantly more probes (t-test, P < 0.05) in the riverine sites communities (22,880 ± 680) than in the backwater sites (18,022 ± 830). The overlap of detected probes within backwater/riverine sites (90.46% ± 3.88%) was much higher than that between samples from backwater and riverine sites (73.26% ± 2.81%, one-way ANOSIM, P = 0.0001). The 74 most abundant functional genes (sum of normalized signal intensities > 100) were all significantly different between the backwater and riverine sites (Table S5). Generally, profiles derived from the riverine sites showed significantly higher gene signal intensities than those derived from the backwater sites (t-test, P < 0.05). Moreover, 72.14% of all detected functional genes were significantly different between the communities derived from backwater and riverine sites, which was confirmed by dissimilarity testing (PERMANOVA, F = 0.807, P = 0.001).
For each gene category (e.g., carbon, nitrogen, phosphorus and sulfur cycling; metal homeostasis; organic remediation; secondary metabolism; and virulence), significantly fewer probes showed positive signals in samples from backwater sites than those from riverine sites (t-test, P < 0.01). Significant differences were further confirmed by comparing normalized signal intensities for each gene category. Similar results were obtained at the sub-category level, with the exception of nitrogen assimilation genes (Table S6). CCA analysis indicated that TP, N-NO3, COD and temperature were significantly associated with microbial functional diversity. The first two axes explained 76.0% of the functional information (Fig. 2B). TP was the most important environmental factor affecting functional diversity (Fig. 3B).
Functional characteristics determined using metagenomic shotgun sequencing
Metagenomic shotgun sequencing generated 211 million raw reads (31–41 million per sample) with a total of 42 Gb for all six samples (6.4–8.1 Gb per sample). Most of reads (96.6%) passed our strict quality control requirements. As sequence depth can affect estimation of the relative abundances of gene categories33, all shotgun datasets were rarefied to the same amount of reads via random resampling before downstream analysis. The valid sequences were assembled into contigs with a total length of 37.1–143.0 Mbp for different samples (N50 = 942 ± 105 bp) and 48,789–219,956 open reading frames (ORFs, > 500 bp) were annotated and matched using the KEGG and eggNOG databases. Finally, a total of 4,024 KEGG orthology (KO) groups involved in various pathways were annotated. Although there were no significant differences (t-test, P > 0.05) in the number of detected ortholog groups, the samples clustered into two groups based on relative abundances (Fig. S3) and indicated clear differences between backwater and riverine sites in the Xiangxi River. The KEGG ortholog groups involved in the carbon, nitrogen and sulfur metabolic pathways showed similar patterns (Fig. S4, S5, S6).
Although only two COG functional categories (cytoskeleton and RNA processing and modification) were significantly different between the backwater and riverine sites (Table S7), in generally most of the categories related to core metabolic functions (e.g., energy production and conversion, amino acid, coenzyme, lipid and nucleotide transport and metabolism, secondary metabolites biosynthesis, transport and catabolism) showed relatively higher abundance in the backwater sites than in the riverine sites.
Discussion
It is well known that plankton are very sensitive to environmental variations and therefore the physical and chemical changes associated with damming may have a detrimental effect on plankton. In addition, since plankton are at the bottom of the food chain, biological impacts of damming will present first effects on the plankton community as shifts in composition or abundance9. Water flow velocity, which is one of the most important factors affecting plankton in river systems3, can be significantly decreased by damming regulation. The Three Gorges Dam, constructed in the middle reach of the Yangtze River from 1994 and 2006, has significantly increased the Xiangxi River water level23 and decreased flow velocity in the main channel7. The effects of the water being held or pushed back (backwater) can be felt more than 30 km up the estuary of Xiangxi River. Six representative sites along this river, representing direct dam effects (i.e., backwater XXR_E, XXR_M and WJB) or just indirect dam influences (i.e., riverine XXR_U, BSR_E and SDR_E, Fig. 1) were studied. We hypothesized that the structural and functional patterns of communities directly impacted by the Three Gorges Dam would be significantly different from those indirectly affected.
16S rRNA gene sequencing results indicated that the backwater area showed higher bacterioplankton α-diversity than riverine sites (Fig. S2). This may be attributed to: (i) a greater abundance of protozoa and rotifer and more zooplankton taxa in the riverine sites (Table S2) exerting a strong top-down effect on the bacterioplankton31,34; and/or (ii) the lake-like conditions in the backwater sites, characterized by low flow velocity, low turbidity and greater nitrogenous nutrients (Table S1), may support a greater variety of bacterioplankton lineages7,9. Similar to the bacterial community composition observed in other rivers9,35, Betaproteobacteria was present at a much higher proportion in upstream waters than in downstream waters, whereas Actinobacteria was at a much higher percentage in backwater sites (Table S4). These results support our hypothesis that the bacterial community composition was significantly impacted by damming and the associated environmental changes. This was further confirmed by the Mantel test (Table 1) and also in agreement with a recently reported study showing that bacterioplankton community structure was affected by a large dam in the Ebro River9.
Next, we wanted to determine whether the compositional differences within the communities inevitably affected community-level function, particularly carbon, nitrogen, phosphorus and sulfur cycling. The functional gene-based GeoChip 5.0 used here detected a higher functional α-diversity than that calculated from OTU composition, suggesting that particular species show distinct functional potential in response to environmental variation. Although there is some debate about the use of functional α-diversity, it has been increasingly applied to both macro- and micro-organisms to evaluate the functional characteristics of targeted communities36,37,38,39. We found riverine communities showed significantly higher functional gene richness than backwater communities (t-test, P < 0.05), which is in almost complete contrast to the bacterioplankton taxonomic richness. The metagenomic KO diversity showed good agreement with the functional gene diversity and was also negatively correlated with taxonomic diversity (Fig. 4). These results suggest that taxonomic and functional analysis should be employed simultaneously for a better understanding of the microbial “black-box”, because community structure does not necessarily dictate functional diversity of a microbial community40.

Pairwise comparison performed based on the α-diversity (Shannon index) determined using metagenomic shotgun sequencing (KO diversity), GeoChip 5.0 (gene diversity) and amplicon sequencing (16S rRNA gene diversity).
The 95% confidence and prediction intervals are given (inner and outer dashed lines, respectively).
Interestingly, 67.0% of the detected genes showed significant relationships to environmental factors, but only 11.2% of the taxonomic genera were significantly correlated with environmental conditions (Table 2). This seems to imply that functional gene patterns may be more sensitive to environmental variations than taxonomic composition. Similarly, functional genes appeared to be more appropriate than ‘species’ information in addressing questions regarding bacterial community assembly16. As bacterioplankton are essential players in the release of phosphorus41 and nitrogen fixation in aquatic ecosystems29, changes in functional genes involved in nitrogen and phosphorus cycling theoretically provide important implications to geochemical cycling. We found that TP and nitrogen sources (N-NH4 and N-NO3) were the most important factors affecting the bacterioplankton community function and composition, respectively (Fig. 3). The relationship between bacterioplankton and TP is frequently observed in freshwater systems28,42. The relationship between taxonomic composition and nitrogen sources is also not surprising as that was reported in some other river system such as Ebro River9. This correlation may be attributed to the higher abundance of Proteobacteria (Table S4), which includes many members involved in nitrogen cycling29,43. There really has high abundance of Proteobacteria OTUs/genera (e.g., Limnohabitans, Acidovorax, Table S3.1, S3.2) involved in nitrogen cycling. However, in-depth studies are needed to determine how the detected bacterial members/genes stimulate nutrient cycling processes in this ecosystem.
The metabolic potential and activity revealed by shotgun sequencing suggest that both the overall metabolic pathway (Fig. S3) and the specific biochemical processes (Fig. S4, S5, S6) reflect correlations between the bacterioplankton community and environmental variations caused be damming. For example, the lower reaches of the Xiangxi River have been transformed from a riverine system into a lacustrine system28,44. The resultant environmental changes have caused water blooms in some of the newly formed bays in the Xiangxi River24,25. The Mantel test further confirmed the significant relationship between KO groups and environmental factors (r = −0.564, P = 0.029, Table 1). Interestingly, 35.48% of the KO groups (Fig. S5, Table 3) involved in nitrogen cycling showed significant differences between the backwater and riverine sites. These significantly different KO groups were primarily involved in coding enzymes that catalyze the conversion of nitrate to ammonia (Fig. S7), which is in agreement with gene abundance data obtained from the GeoChip 5.0 (Fig. S8). The lower nitrogenous nutrients in the riverine sites (Table S1) could be associated with the higher abundance of algae, as nitrogen has been found to be a limiting nutrient for phytoplankton growth in the Xiangxi River45. The higher abundance of phytoplankton in the riverine sites during our investigating period (as determined by morphological counting and the concentration of Chl a) possibly consumed considerable nitrogenous nutrients and therefore the concentrations of N-NH4, N-NO3 and TN were relatively low. This high nitrogenous requirement of phytoplankton may stimulate the bacteria to try and regenerate these nutrients by increasing populations containing the functional genes involved in nitrogen cycling, resulting in higher signal intensities of those genes in the riverine sites (Fig. S8). We should also acknowledge that there may have some other biogeochemical processes contributing to the differences of detected nitrogenous substances.
In summary, our results suggest that the Three Gorges Dam does significantly affect the structure and function of bacterioplankton communities in the Xiangxi River as shown by clear community patterns between sites with or without direct dam effects, which represented by backwater and riverine communities, respectively (Table 3). In addition, although the different methods used in describing the microbial diversity did not necessarily give identical results33,46, our results suggest that the three important culture-independent approaches used in this study detected similar bacterioplankton patterns along the Xiangxi River. Moreover, all of the diversity revealed by the different methods significantly correlated with the environmental factors. The nearest taxon index (NTI, all values larger than 2) also confirmed that environmental filtering was one of the major processes driving the bacterioplankton community changes in the Xiangxi River. This study not only provides a comprehensive metagenomic insight into the bacterioplankton community in the well-known Three Gorges Reservoir, but also enhances our understanding on the effects of damming on microbial community function.
Methods
Study area and sampling procedures
This study was conducted in the Xiangxi River, which lies 32 km upstream of the Three Gorges Dam, has a mainstream length of 94 km, a watershed area of 3,099 km2 and a natural fall of 1,540 m47. To address the possible impacts of the Three Gorges Dam on the bacterioplankton communities, we collected the samples from the estuary (XXR_E), midstream (XXR_M) and upstream of Xiangxi River (XXR_U), as well as Wujia Bay (WJB) and two secondary tributary sites at the estuary of Baisha River (BSR_E) and Shendu River (SDR_E) in April 2013 before the spring bloom. These sites can be classified into backwater (XXR_E, XXR_M and WJB) and riverine sites (XXR_U, BSR_E and SDR_E, Fig. 1). They were selected to represent a wide range of environmental gradients under the influences of the Three Gorges Dam, which may harbor various bacterioplankton communities and can be used to further determine how the functional capabilities are affected by a huge dam. In this study, we only examined bacterioplankton at a single time point per site, as it was not our goal to evaluate temporal variability.
At each site, equal volumes of water (10 L) were collected from the depths of 0.5, 5 and 10 m in the middle point of the river. After mixed fully in a bucket, water was split for the following different experiments. For DNA-based analysis, one liter of this mixed water was sequentially filtered through a 1.2-μm (Whatman, NJ, USA) and 0.22-μm filter paper (Millipore, MA USA) to collect most of the bacterioplankton organisms. The filtration was immediately performed onboard and the filters were stored at −20°C until DNA extraction. Samples for morphological analysis were fixed with 1.5% Lugol's solution (final concentration) and after 24 h sedimentation, the above solution was removed by a siphon and the microplankton that had settled to the bottom were concentrated to 30 mL for abundance determination. Another set of samples was pre-processed based on standard methods48 for physicochemical analysis.
Physicochemical analysis and morphological identification
Conductivity, dissolved oxygen (DO), turbidity and pH were measured using a Professional Plus Multi-Parameter Probe (YSI, OH, USA) in situ. Temperature was read from a thermometer and transparency was determined using a Secchi disc. Chemical characteristics, including chemical oxygen demand (COD), oxidation-reduction potential (ORP), total nitrogen (TN), ammonium nitrogen (N-NH4), nitrate nitrogen (N-NO3), total phosphorus (TP) and phosphate phosphorus (P-PO4) were measured using standard methods as described previously48. Chlorophyll a (Chl a) was extracted from a Whatman GF/C filter for 24 h with 90% acetone, centrifuged at 3,000 rpm for 10 min and then spectrophotometrically quantified48.
Zooplankton were identified and counted under an Axioplan 2 Imaging microscope (Zeiss, Jena, Germany) using a method described previously4. Phytoplankton analysis was performed under an optical microscope (Olympus BH-2, Tokyo, Japan) as previously described25.
DNA extraction and GeoChip 5.0 analysis
The 1.2-μm and 0.22-μm filters used to collect microorganisms were used to extract DNA with the PowerWater® DNA Extraction kit (Mo Bio, CA, USA) separately according to the manufacturer's instructions. The DNA extracted from the two filters were pooled as a single sample and purified using a Genomic DNA Clean & Concentrator™ kit (Zymo, CA, USA) for downstream experiments. The purified DNA concentration was quantified using a PicoGreen dsDNA Assay kit (Invitrogen, CA, USA).
For each sample, 500 ng of DNA was labeled with the fluorescent dye Cy-3 (GE Healthcare, CA, USA) by random priming as described previously49; then purified using a QIAquick Purification kit (Qiagen, CA, USA) and then dried in a SpeedVac (Thermo Savant, NY, USA). The dried DNA was rehydrated with 13 μL of DNase/RNase-free distilled water, mixed completely and then centrifuged to collect all liquid at the bottom of the tube. A total of 42 μL of buffer, including 1× HI-RPM hybridization buffer,1× aCGH blocking agent, 0.05 μg·μL−1 Cot-1 DNA, 10 pM universal standard and 10% formamide (final concentrations), was added to each sample. After mixing completely, the solution was spun down and incubated at 95°C for 3 min, then incubated at 37°C for 30 min. The prepared samples were hybridized with GeoChip 5.0 arrays (60 K) at 67°C for 24 h with a rotation at 20 rpm in a hybridization oven. GeoChip 5.0 is the newest generation of GeoChip. The 60 K arrays used in this study contained approximately 60,000 probes targeting hundreds of gene families involved in various biogeochemical processes such as carbon, nitrogen, phosphorus and sulfur cycling, metal homeostasis, organic remediation, secondary metabolism and virulence. The scanned images of hybridized GeoChips were converted and extracted using Agilent Feature Extraction 11.5 software (Agilent Technologies, Inc., CA, USA); the extracted information was then analyzed using the microarray analysis pipeline on our web site (http://ieg.ou.edu/microarray/) as previously described50.
MiSeq sequencing of 16S rRNA gene amplicons
The composition of bacterioplankton was analyzed using Illumina MiSeq sequencing of 16S rRNA gene amplicons. The V4 region of the 16S rRNA gene, which can yield accurate taxonomic information and shows few biases for various bacterial taxa51, was amplified with the primer set 515f (5′-GTGCCAGCMGCCGCGGTAA-3′)/806r 5′-GGACTACHVGGGTWTCTAAT-3′) and all PCR amplifications were conducted in triplicate for each sample. An initial 10 cycles of PCR amplification were performed. The products were then purified with Agencourt® Ampure® XP (Beckman Coulter, Inc., CA, USA) and used as a template for the second PCR amplification of 20 cycles using the same primer set; however, the reverse primer contained the appropriate adapters and different barcodes to distinguish samples. PCR products were visualized using 1% agarose gels stained with ethidium bromide and negative controls were always performed to confirm the absence of contamination. True positive amplicons were quantified using a PicoGreen dsDNA Assay kit (Invitrogen, CA, USA), combined equally and then gel purified. The DNA library was sequenced at the Institute for Environmental Genomics using the Illumina MiSeq platform according to the manufacturer's instructions.
Quality filtering and the processing of MiSeq reads were conducted on our Galaxy pipeline. After trimming the primers and deleting sequences containing Ns, high quality sequences in length of 245–260 bp were kept for subsequent analysis. We ultimately obtained 18,256–24,012 high-quality sequences for different samples. To correct for differences in sequencing depth, 17,570 OTUs were randomly resampled for each sample before performing the downstream analysis. The taxonomic dissimilarities/similarities between each pair of communities were determined using the Bray-Curtis and Raup-Crick indexes.
Metagenomic shotgun sequencing, assembly and pathways analysis
For each sample, 3 μg of DNA was placed in the shotgun sequencer at the Novogene Bioinformatics Institute according to the manufacturer's established protocols. Briefly, the DNA was broken into fragments using the Covaris™ S220 System (Applied Biosystems, CA, USA) and extracted using the QIAquick PCR Purification kit (Qiagen, Hilden, Germany). Libraries were prepared following a standard protocol from Illumina and 100-bp paired-end reads were sequenced using the Illumina HiSeq 2000 platform according to the manufacturer's instructions. The metagenomic datasets are publicly available in the MG-RAST system (http://metagenomics.anl.gov/) under project identifiers from 4543171.3 to 4543182.3.
After quality filtering as described elsewhere33, all shotgun metagenomic datasets were rarefied to the same sequencing depth by random resampling 6,144,417,000 bp valid sequences for each sample before downstream analysis. The valid sequences were assembled using SOAPdenovo V 1.0652 with the parameters -K 27 (k-mer size)-R-M3-d1. Genes were predicted using MetaGeneMark53 and BLASTP54 was used to search the protein sequences of the predicted genes within the eggNOG database55 and KEGG database56 with E ≤ 1 × 10−5. The genes were annotated using the eggNOG or KEGG homologs with the lowest e-value. To construct the metabolic pathways associated with the bacterioplankton organisms, sequences were mapped using USEARCH57 with E ≤ 1 × 10−5 against the KO database. The significant hits were imported into HUMAnN58 and the abundance and coverage were calculated for each KEGG metabolic pathway. The abundance data were also analyzed for complete linkage clustering based on the Euclidean distance.
Statistical analysis
The three datasets generated by 16S rRNA gene sequencing (OTU composition), GeoChip analysis (functional gene signal intensities) and metagenomic shotgun sequencing (relative abundance of KO groups) were further analyzed with one or all of the following statistical methods: (i) α-/β-diversity comparison and response ratio analysis; (ii) clustering based on the community structural or functional characteristics; (iii) dissimilarity test by permutational multivariate analysis of variance (PERMANOVA) with Bray-Curtis dissimilarity for comparing each dataset or sub-dataset; (iv) significance tests based on unpaired Student's t-test to identify differences between any two compared objects; (v) CCA and partial CCA to determine the community variation that can be explained by the environmental variables; and (vi) Mantel test to determine the relationship between any two of the targeted matrices. All statistical analyses described above were performed using the R package vegan (R Foundation for Statistical Computing, Vienna, Austria) or our R-based pipeline (http://ieg.ou.edu/microarray/). The analysis of ANOSIM and Raup-Crick were performed by using the software of Past.
References
Karner, M. B., DeLong, E. F. & Karl, D. M. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409, 507–510 (2001).
Woese, C. R. There must be a prokaryote somewhere: microbiology's search for itself. Microbiol. Rev. 58, 1–9 (1994).
Crump, B. C. & Hobbie, J. E. Synchrony and seasonality in bacterioplankton communities of two temperate rivers. Limnol. Oceanogr. 50, 1718–1729 (2005).
Yu, Y. H., Yan, Q. Y. & Feng, W. S. Spatiotemporal heterogeneity of plankton communities in Lake Donghu, China, as revealed by PCR-denaturing gradient gel electrophoresis and its relation to biotic and abiotic factors. FEMS Microbiol. Ecol. 63, 328–337 (2008).
Comte, J. & del Giorgio, P. A. Linking the patterns of change in composition and function in bacterioplankton successions along environmental gradients. Ecology 91, 1466–1476 (2010).
Helland-Hansen, E., Holtedahl, T. & Liye, K. A. Hydropower Development, Vol. 3: Enviromental Effects (Norwegian Institute of Technology Press, Trondheim, 1995).
Dumestre, J. F., Casamayor, E. O., Massana, R. & Pedrós-Alió, C. Changes in bacterial and archaeal assemblages in an equatorial river induced by the water eutrophication of Petit Saut dam reservoir (French Guiana). Aquat. Microb. Ecol. 26, 209–221 (2001).
Wang, S. et al. Diversity of microbial plankton across the Three Gorges Dam of the Yangtze River, China. Geosci. Front. 3, 335–349 (2012).
Ruiz-González, C., Proia, L., Ferrera, I., Gasol, J. M. & Sabater, S. Effects of large river dam regulation on bacterioplankton community structure. FEMS Microbiol. Ecol. 84, 316–331 (2013).
López-García, P., Rodríguez-Valera, F., Pedrós-Alió, C. & Moreira, D. Unexpected diversity of small eukaryotes in deep-sea Antarctic plankton. Nature 409, 603–607 (2001).
Moon-van der Staay, S. Y., De Wachter, R. & Vaulot, D. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409, 607–610 (2001).
Zhou, J. Z. et al. Bacterial phylogenetic diversity and a novel candidate division of two humid region, sandy surface soils. Soil Biol. Biochem. 35, 915–924 (2003).
Tringe, S. G. & Rubin, E. M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 6, 805–814 (2005).
Tringe, S. G. et al. Comparative metagenomics of microbial communities. Science 308, 554–557 (2005).
Steffen, M. M. et al. Comparative metagenomics of toxic freshwater cyanobacteria bloom communities on two continents. PLoS ONE 7, e44002 (2012).
Burke, C., Steinberg, P., Rusch, D., Kjelleberg, S. & Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. U. S. A. 108, 14288–14293 (2011).
Davenport, C. F. & Tümmler, B. Advances in computational analysis of metagenome sequences. Environ. Microbiol. 15, 1–5 (2013).
He, Z. L., Deng, Y. & Zhou, J. Z. Development of functional gene microarrays for microbial community analysis. Curr. Opin. Biotech. 23, 49–55 (2012).
He, Z. L., Van Nostrand, J. D. & Zhou, J. Z. Applications of functional gene microarrays for profiling microbial communities. Curr. Opin. Biotech. 23, 460–466 (2012).
Xie, W. et al. Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J. 5, 414–426 (2011).
Oh, S. et al. Metagenomic insights into the evolution, function and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl. Environ. Microbiol. 77, 6000–6011 (2011).
Ghai, R. Metagenomics of the water column in the pristine upper course of the Amazon river. PLoS ONE 6, e23785 (2011).
Wang, H. Y. Effects of the Three Gorges Reservoir on the water environment of the Xiangxi River with the proposal of countermeasures. Resour. Environ. Yangtze Basin. 14, 233–237 (2005).
Ye, L., Xu, Y. Y., Han, X. Q. & Cai, Q. H. Daily dynamics of nutrients and chlorophyll a during a spring phytoplankton bloom in Xiangxi Bay of the Three Gorges Reservoir. J. Freshwater Ecol. 21, 315–321 (2006).
Zhou, G. J. et al. Phytoplankton variation and its relationship with the environment in Xiangxi Bay in spring after damming of the Three-Gorges, China. Environ. Monit. Assess. 176, 125–141 (2011).
Bi, Y. H. et al. The effect of TGR's experimental impoundment on phytoplankton communities in Xiangxi River, China. Phycologia 48, 8–8 (2009).
Sekiguchi, H., Watanabe, M., Nakahara, T., Xu, B. H. & Uchiyama, H. Succession of bacterial community structure along the Changjiang River determined by denaturing gradient gel electrophoresis and clone library analysis. Appl. Environ. Microbiol. 68, 5142–5150 (2002).
Yan, Q. Y., Yu, Y. H., Feng, W. S., Yu, Z. G. & Chen, H. T. Plankton community composition in the Three Gorges Reservoir Region revealed by PCR-DGGE and its relationships with environmental factors. J. Environ. Sci. 20, 732–738 (2008).
Yang, X. et al. Decrease of NH4+-N by bacterioplankton accelerated the removal of cyanobacterial blooms in aerated aquatic ecosystem. J. Environ. Sci. 25, 2223–2228 (2013).
Raup, D. M. & Crick, R. E. Measurement of faunal similarity in paleontology. J. Paleontol. 53, 1213–1227 (1979).
Jürgens, K. & Matz, C. Predation as a shaping force for the phenotypic and genotypic composition of planktonic bacteria. Anton. Leeuw. Int. J. G. 81, 413–434 (2002).
Belasky, P. Biogeography of Indo-Pacific larger foraminifera and scleractinian corals: a probabilistic approach to estimating taxonomic diversity, faunal similarity and sampling bias. Paleogeogr. Paleoclimatol. Paleoecol. 122, 119–141 (1996).
Fierer, N. et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. U. S. A. 109, 21390–21395 (2012).
Šimek, K. et al. Shifts in bacterial community composition associated with different microzooplankton size fractions in a eutrophic reservoir. Limnol. Oceanogr. 44, 1634–1644 (1999).
Warkentin, M., Freese, H. M. & Schumann, R. Bacterial activity and bacterioplankton diversity in the eutrophic River Warnow: direct measurement of bacterial growth efficiency and its effect on carbon utilization. Microb. Ecol. 61, 190–200 (2011).
Diaz, S. & Cabido, M. Vive la différence: plant functional diversity matters to ecosystem processes. Trends Ecol. Evol. 16, 646–655 (2001).
Petchey, O. L. & Gaston, K. J. Functional diversity (FD), species richness and community composition. Ecol. Lett. 5, 402–411 (2002).
Green, J. L., Bohannan, B. J. M. & Whitaker, R. J. Microbial biogeography: from taxonomy to traits. Science 320, 1039–1043 (2008).
Raes, J., Letunic, I., Yamada, T., Jensen, L. J. & Bork, P. Toward molecular trait-based ecology through integration of biogeochemical, geographical and metagenomic data. Mol. Syst. Biol. 7, 473 (2011).
Frossard, A., Gerull, L., Mutz, M. & Gessner, M. O. Disconnect of microbial structure and function: enzyme activities and bacterial communities in nascent stream corridors. ISME J. 6, 680–691 (2012).
Wang, Y. Y. & Chen, F. Z. Decomposition and phosphorus release from four different size fractions of Microcystis spp. taken from Lake Taihu, China. J. Environ. Sci. 20, 891–896 (2008).
Artigas, J. et al. Phosphorus use by planktonic communities in a large regulated Mediterranean river. Sci. Total. Environ. 426, 180–187 (2012).
Breitbart, M. et al. Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Ciénegas, Mexico. Environ. Microbiol. 11, 16–34 (2009).
Zeng, H., Song, L. R., Yu, Z. G. & Chen, H. T. Distribution of phytoplankton in the Three-Gorge Reservoir during rainy and dry seasons. Sci. Total. Environ. 367, 999–1009 (2006).
Zhou, G. J., Bi, Y. H., Zhao, X. M., Chen, L. & Hu, Z. Y. Algal growth potential and nutrient limitation in spring in three-Gorges reservoir, China. Fresen. Environ. Bull. 18, 1642–1647 (2009).
Steven, B., Gallegos-Graves, L., Starkenburg, S. R., Chain, P. S. & Kuske, C. R. Targeted and shotgun metagenomic approaches provide different descriptions of dryland soil microbial communities in a manipulated field study. Env. Microbiol. Rep. 4, 248–256 (2012).
Wang, J., Wang, B. S. & Luo, Z. Q. Dictionary of the Yangtze River (Wuhan Press, Wuhan, 1997).
Huang, X. F. Survey, Observation and Analysis of Lake Ecology (China Standard Press, Beijing, 2000).
He, Z. L. et al. Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2 . Ecol. Lett. 13, 564–575 (2010).
Yang, Y. F. et al. Responses of the functional structure of soil microbial community to livestock grazing in the Tibetan alpine grassland. Global Change Biol. 19, 637–648 (2013).
Bates, S. T. et al. Examining the global distribution of dominant archaeal populations in soil. ISME J. 5, 908–917 (2011).
Li, R. Q. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Zhu, W. H., Lomsadze, A. & Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, e132 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Muller, J. et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 38, D190–D195 (2010).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 (2008).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Abubucker, S. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comp. Biol. 8, e1002358 (2012).
Acknowledgements
This work was supported by the China Three Gorges Corporation (IHB/CN/2012103), the Youth Innovation Promotion Association, CAS (Y22Z07) and the National Natural Science Foundation of China (31071896).
Author information
Authors and Affiliations
Contributions
Q.Y., Y.B., Z.H. and Y.Y. conceived the research. Q.Y., Y.B. and J.L. performed the experiments. Q.Y. wrote the manuscript. Y.B., Y.D., Z.H., J.D.V.N. and J.Z. edited the manuscript. X.W., L.W., Y.D. and Z.S. contributed sampling, reagents or data analysis pipeline. All authors reviewed and accepted the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yan, Q., Bi, Y., Deng, Y. et al. Impacts of the Three Gorges Dam on microbial structure and potential function. Sci Rep 5, 8605 (2015). https://doi.org/10.1038/srep08605
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08605
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
