
Abstract
Magnetic resonance (MR) methods to detect and quantify fluorine (19F) nuclei provide the opportunity to study the fate of cellular transplants in vivo. Cells are typically labeled with 19F nanoparticles, introduced into living organisms and tracked by 19F MR methods. Background-free imaging and quantification of cell numbers are amongst the strengths of 19F MR-based cell tracking but challenges pertaining to signal sensitivity and cell detection exist. In this study we aimed to overcome these limitations by manipulating the aminophospholipid composition of 19F nanoparticles in order to promote their uptake by dendritic cells (DCs). As critical components of biological membranes, phosphatidylethanolamines (PE) were studied. Both microscopy and MR spectroscopy methods revealed a striking (at least one order of magnitude) increase in cytoplasmic uptake of 19F nanoparticles in DCs following enrichment with 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE). The impact of enriching 19F nanoparticles with PE on DC migration was also investigated. By manipulating the nanoparticle composition and as a result the cellular uptake we provide here one way of boosting 19F signal per cell in order to overcome some of the limitations related to 19F MR signal sensitivity. The boost in signal is ultimately necessary to detect and track cells in vivo.
Similar content being viewed by others
Introduction
Tracking immune cells in vivo is a prerequisite for understanding the development of pathologies associated with disorders of the immune system1. Dendritic cells (DCs) are immune cells that play key roles in the development of immunity and immunopathology. Of note, an understanding of the distribution and fate of cells such as DCs following their therapeutic application in vivo such as in cancers is crucial to assess treatment efficacy2. The lack of a priori knowledge of the kinetics and dynamics of these cells during physiological and pathological settings makes their localization a challenging task. It is therefore a top priority to develop methods for the non-invasive spatiotemporal tracking of immune cells in vivo that can be easily transferred to the clinical scenario3,4,5,6.
Magnetic resonance imaging (MRI) offers an ideal solution for tracking cells in vivo due to its non-invasiveness and clinical translation as well as the opportunity of repetitive measurements and longitudinal studies. Major challenges in differentiating cells from the recipient tissue and signal sensitivity constraints nevertheless exist. For instance, iron oxide nanoparticles that reduce T2* relaxation7 in MR have been used to label cells, however present a challenge whereby the contrast created by the labeled cells is not easily distinguishable from other intrinsic tissue contrasts8. This limitation of cellular MRI is surmounted by fluorine (19F) MR techniques3. Carbon-bound fluorine is absent in living organisms. This guarantees background-free MR signals for externally-applied 19F compounds. Therefore 19F MR techniques are advantageous for localizing 19F-containing exogenous agents in vivo since they permit complete signal selectivity and specificity9,10,11. Importantly, the 19F MR signal can also be equated to measurable cell numbers within defined regions6,12. For in vivo tracking, cells are typically labeled with nanoparticles enriched with perfluorocompounds (PFCs) prior to their introduction into living organisms3,4,5,6. These compounds possess unique properties (inertness, biocompatibility and hydrophobicity) which stem from the C–F bonds within the molecular structure13. Hydrophobicity becomes more significant with increasing number of 19F atoms and a critical factor for increasing the ability of these compounds to cross biological membranes14,15. Making use of these properties, 19F-rich nanoparticles are prepared via various techniques – commonly by emulsifying PFCs with phospholipids or surfactants – in order to label and track immune cells in vivo with the aid of combined 19F and anatomical/proton (1H) MR imaging as well as 19F spectroscopy techniques3,4,5,6. Notwithstanding the benefits specified above, some limitations and challenges exist for 19F-based MR techniques, especially those pertaining to detection limit and signal sensitivity. These limitations come at a cost in increased signal averaging and thus acquisition times to compensate for low signal-to-noise ratio (SNR). There is a need to optimize cellular 19F MR methods especially since clinical trials with DC vaccines have thus far provided a proof-of-principle as cancer therapy16 and the applicability of 19F MR techniques for DC vaccines are being explored in colorectal cancer patients17.
In the present study we explored the possibility of altering the composition of 19F nanoparticles in order to maximize their uptake by DCs and therefore to promote the 19F signal per cell. To meet this goal, we investigated the incorporation of phosphatidylethanolamines (PEs) into the 19F nanoparticle shell. PEs are aminophospholipids that constitute an integral part of biological membranes18,19. These aminophospholipids are cone-shaped, they do not form bilayers but inverted hexagonal phases, are believed to exert a lateral pressure that regulates membrane curvature and, are thought to stabilize membrane proteins in their optimum conformations20. PE analogs have been used successfully in non-viral transfection systems21,22 as well as in nanotube carriers for drug delivery23. For all these reasons we studied the influence of PE enrichment on the uptake of 19F nanoparticle by DCs. Using both electron microscopy and 19F MR spectroscopy we observed a dramatic increase in uptake of 19F nanoparticles enriched with 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) by DCs. As a result, the 19F signal/cell was increased by at least one order of magnitude and the cell detection limit considerably reduced. With this advantage at hand we could reduce the PFCE concentration per nanoparticle to avoid impairment in cell function, particularly cell migration. Our findings further offset the constraints of 19F MR and bring us a step closer to the crucial goal of ultimate signal sensitivity and minimal cell detection limit.
Results
Dipalmitoyl-Phosphoethanolamine promotes nanoparticle uptake by DCs
Considering the power of PE analogs to promote cellular uptake in transfection and drug delivery systems21,22,23, we first set off to enrich 19F-rich (PFCE) nanoparticles (NP) used in our previous studies5,6,12 with different phospholipids of the phosphoethanolamine (PE) family (Figure 1A). The basic nanoparticles were prepared using Pluronic F-68 block copolymer. To enrich the 19F nanoparticles with PE we chose two analogs differing in their two long fatty acid hydrocarbon chains; one PE contained one unsaturated bond (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, POPE) and the other PE consisted of only saturated bonds (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine, DPPE). Also consisting solely of saturated bonds was the third candidate we chose: 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), bound to polyethylene glycol-2000 (DSPE-PEG2000). PEG is widely employed as polymeric steric stabilizer and is anchored to stealth liposomal surfaces via cross-linked DSPE lipid24,25. PEG2000 was chosen since blocks of higher molecular weight in similarly-sized nanoparticles have been shown to reduce nanoparticle uptake in macrophages (uptake: PEG2000 > PEG5000 > PEG10000)26. We used DPPE and POPE to enrich Pluronic-based PFCE-containing nanoemulsions and prepared liposomal PFCE nanoparticles using DSPE-PEG2000. For all nanoparticle preparations, the molar fraction of the constituents and the physical characteristics (size, polydispersity, surface charge) are shown in Table 1.

Selection of phosphoethanolamines for enrichment of PFCE nanoparticles.
(a) Chemical structures of DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine), POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine used for the 19F nanoemulsion and DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N[methoxy(polyethylene glycol) -2000]) used for the liposomal nanoparticles. (b) DCs were labeled with different 19F nanoparticle preparations (DSPE-PEG2000, Pluronic-basic, Pluronic-POPE and Pluronic-DPPE) using a PFCE concentration of 20 μmol per 107, fixed in 2% PFA and transferred (106) to NMR tubes. After positioning in a 19F-tuned loop coil (see Methods), 19F signal was acquired using a 90° block excitation pulse with 10 kHz bandwidth and the PFCE amount per 106 calculated using a 500 mM PFCE standard.
To determine differences in uptake between the different nanoparticle groups we performed 19F magnetic resonance spectroscopy (MRS) of the fixed DCs following labeling to determine the amount of 19F compound (PFCE) per 106 cells. Pluronic-nanoparticles enriched with PE polymers appeared to be taken up more efficiently by DCs than basic Pluronic nanoparticles and DSPE–PEG2000 liposomes as determined by the increase in cellular 19F signal. POPE enrichment already resulted in an increased 19F signal (POPE 1x: 138 nmol per 106 cells and POPE 10x: 179 nmol per 106 cells) compared to the basic nanoparticle formulations (74 nmol per 106 cells) and DSPE–PEG2000 liposomes (69 nmol per 106 cells). When we employed DPPE to enrich the nanoparticle shell, we observed an even stronger enhancement of 19F signal (771 nmol per 106 cells) (Figure 1B). This equates to an increase in 19F spins from 0.89 × 1012 19F spins (in basic formulations) to 0.93 × 1013 19F spins (in DPPE-enriched nanoparticles) per dendritic cell unit.
Intracellular appearance of Dipalmitoyl-Phosphoethanolamine-enriched nanoparticles
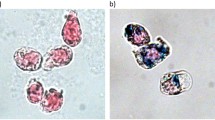
Despite the dramatic differences in 19F signal within DCs between the different nanoparticles, we did not observe any conspicuous differences in the shape of the nanoparticles employed as investigated by Cryo-TEM (Figure 2A). We next went on to investigate whether the striking increase in 19F signal in the DPPE-enriched nanoparticles was the result of increased nanoparticle uptake. In ultrathin sections of DCs derived from the same culture conditions, we observed intense differences in cytoplasmic uptake between the nanoparticle formulations (Figure 2B, EM images in upper two panels and lower left panel). We commonly observed the nanoparticles as white globules within the cell cytoplasm but similar to our previous observations5 the nanoparticles also often appeared compartmentalized as clusters in a lipid membrane capsule within an amorphous grey compartment. We also observed increased DC uptake of nanoparticles enriched with DPPE linked to the tracer dye Rhodamine as shown from the Rhodamine fluorescence imaged by laser scanning microscopy (Figure 2B, LSM image in lower right panel); these nanoparticles were also loaded into DCs similarly to the non-Rhodamine linked DPPE-enriched 19F nanoparticles.

Appearance of phosphoethanolamine-enriched PFCE nanoparticles.
(a) Cryogenic transmission electron microscopy (TEM) images of DSPE-PEG2000, Pluronic-basic, Pluronic-DPPE and Pluronic-DPPE-Rhodamine nanoparticles encapsulating PFCE fluorine compound (size-bar for TEM images: 50 nm). (b) Upper two panels and lower left panel show ultrathin sections of DCs labeled with DSPE-PEG2000, basic and DPPE-enriched PFCE nanoparticles (size-bar for EM images: 2 μm). Lower right panel shows a laser scanning microscopy image of DCs labeled with DPPE-Rhodamine-enriched 19F nanoparticles (size-bar for LSM image: 10 μm). (c) DCs were labeled with different nanoparticle preparations (DSPE-PEG2000 NP, basic NP, DPPE-NP and DPPE-Rhodamine-NP) using a PFCE concentration of 10 μmol per 107, fixed in 2% PFA and transferred (106) to NMR tubes. 19F signal was acquired using a 90° block excitation pulse and the PFCE amount per 106 calculated using a 500 mM PFCE standard.
As a next step, the same groups of cells labeled with basic (-), DPPE or DPPE-Rhodamine 19F nanoparticles as well as DSPE-PEG2000 liposomes were investigated by 19F MRS (Figure 2C). Similarly to the results in Figure 1B, we observed a significant increase in 19F signal per cell when DCs were labeled with nanoparticles enriched with DPPE (361 nmol per 106 cells ≡ 0.44 × 1013 19F spins per cell) compared to the basic formulation (25 nmol per 106 cells ≡ 0.3 × 1012 19F spins per cell) and DSPE–PEG2000 liposomes (35 nmol per 106 cells ≡ 0.42 × 1012 19F spins per cell) (Figure 2C). The 19F signal per cell in DCs labeled with DPPE-Rhodamine nanoparticles was slightly lower (264 nmol per 106 cells ≡ 0.32 × 1013 19F spins per cell) than those from DCs labeled with nanoparticles enriched with DPPE containing no Rhodamine (Figure 2C).
Titrating the 19F label in DPPE-enriched nanoparticles
In the next experiments we performed dose titration curves for the 19F label (perfluoro-15-crown-5-ether, PFCE) in cells loaded with DPPE or basic 19F nanoparticles. With these experiments we wanted to determine the lowest concentration of PFCE feasible for detecting a sufficient 19F signal in MRS (Figure 3). Using the Pluronic-based nanoparticles we had previously employed up to 40 μmol PFCE per 107 DCs in culture5. Taking into account the dramatic increase in 19F signal achieved following enrichment with DPPE we titrated the PFCE amount from 40 μmol to 2 μmol PFCE per 107 DCs. In line with the previous results (Figure 1,2), 5 μmol PFCE label within DPPE 19F nanoparticles gave a larger 19F signal than 40 μmol PFCE label within basic 19F nanoparticles when used to label 107 DCs (Figure 3A, 3B). This observation can be made already from the spectral representations of the 19F signal (Figure 3A) and upon quantification of the 19F signal from the maximum signal intensity of the FID (Figure 3B). By extrapolating the linear fit ([PFCE]label/[PFCE]intracellular) for the basic 19F nanoparticles, we estimate a requirement of 70 μmol for the basic 19F nanoparticles to reach the equivalent 19F signal of DCs labeled with 5 μmol DPPE-enriched nanoparticles (Δ PFCE concentration ~65 μmol) (Figure 3B).

Dose titration of the 19F label for 19F NMR signal.
(a) Spectral representation of 19F signal acquired from basic and DPPE-enriched label within DCs using different doses of the 19F label PFCE. DCs were labeled with nanoparticle preparations using PFCE concentrations between 2 μmol and 40 μmol) per 107, fixed in 2% PFA and transferred (106) to NMR tubes. 19F signal was acquired using a 90° block excitation pulse and spectral representation using a FFT of the acquired FID. (b) The 19F signal was quantified from the intercept of the FID fit and the PFCE amount per 106 was calculated using a 500 mM PFCE standard.
Higher concentrations of 19F label hinder DC migration
Ultimately, the 19F-labeled DCs will be applied in living organisms. Therefore it was necessary to determine the influence of increasing PFCE labeling on DC migration for both DPPE-enriched as well as basic 19F nanoparticles. For this, we employed an agarose assay to determine the chemotaxis of DCs towards a chemokine gradient following maturation with bacterial lipopolysaccharide (LPS). In the initial experiments comparing 19F signal between basic and DPPE nanoparticles we employed 10–20 μmol of PFCE to label 107 cells (Figure 1,2). When we investigated the influence of 10 μmol PFCE label on DC migration, we observed differences in the number of DCs moving towards chemokine between unlabeled cells treated for 18 h with LPS and cells labeled with 19F nanoparticles (Figure 4A). For DCs labeled with DPPE 19F nanoparticles, quantities of PFCE higher than 10 μmol per 107 cells reduced the migration of DCs towards chemokine by more than 50% (Figure 4B). For lower doses of PFCE – specifically 5 μmol per 107 cells – the reduction in migration was less extensive and the difference between DPPE and basic 19F nanoparticles less substantial; the inhibition in migration for DPPE-NP labeled cells was 35% (n = 1270) and the inhibition in migration for basic-NP labeled DCs was 22% (n = 1555) in comparison to unlabeled DCs (n = 1985) (Figure 4B). This finding and considering our previous observation that 5 μmol PFCE in DPPE nanoparticles generate an equivalent 19F signal by at least one order more PFCE in basic nanoparticles (Figure 3B), led us to employ 5 μmol PFCE for all the following experiments.

Dose titration of the 19F label for chemotaxis assay.
(a) DCs (105) labeled with different nanoparticle preparations (10 μmol PFCE) were introduced to glass dishes containing CCL21-agarose spots and the number of cells entering the agarose spot was visualized by acquisition of microscope images. (b) DCs (105) labeled with increasing PFCE concentrations of both basic and DPPE-enriched 19F nanoparticles were introduced to glass dishes containing CCL21-agarose spots and the number of cells entering the agarose spot was determined by acquiring and fusing the microscope images for all the fields of views (FOV) and counting all cells entering the corresponding spot. This experiment is representative of 3.
Lower number of cells detected following labeling with DPPE-enriched nanoparticles
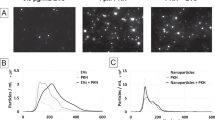
Using 5 μmol PFCE for both DPPE and basic 19F nanoparticles we next measured the intracellular 19F signal in increasing numbers of DCs to determine the cell detection limit. We measured the 19F signal in each fixed cell sample by performing global spectroscopy of the NMR tube containing the cell pellet (see Methods). By using a 19F standard (500 mM PFCE) we quantified the amount of PFCE within each sample. Both spectral representation of the 19F signal (Figure 5A) as well as PFCE quantification (Figure 5B) for both DPPE-enriched and basic 19F nanoparticles demonstrated that 19F signal amplitude correlates with the number of labeled cells.

Lower number of cells detected by 19F MRS following labeling with DPPE-enriched nanoparticles.
(a) Spectral representation of 19F signal acquired from different numbers of basic and DPPE-NP labeled DCs using 5 μmol PFCE per 107 cells for 19F labeling. 19F spectra are represented from the FFT of the acquired FID. (b) The 19F signal was quantified from the intercept of the FID fit and the PFCE amount per 106 was calculated using a 500 mM PFCE standard.
From the spectral representation of the cell dilution curves it was already evident that far less DPPE-NP labeled DCs could be detected than basic-NP labeled DCs (Figure 5A). It should be noted that in these experiments (in line with the chemotaxis assays above) we used low PFCE labeling doses (5 μmol per 107 cells) in contrast to previously published work where we commonly employed 20 μmol per 107 cells5,6,12. In the present experiments we observed that a minimum of 106 DCs labeled with basic nanoparticles (5 μmol) were required to achieve a detectable 19F signal. However for DPPE-NP labeled DCs, 1.5 × 105 of cells could be detected (Figure 5A). Following quantification of the 19F signal from the FID fit and calculation of the intracellular PFCE amount (using PFCE standard), we observed that 105 DPPE-NP labeled DCs give a similar 19F signal and contain similar PFCE amounts (14.48 nmol) as 106 basic-NP labeled DCs (10.45 nmol). It can also be deduced from the spectral representations (Figure 5A) that a baseline intracellular value of approximately 10 nmol PFCE (Figure 5B) is the threshold above which DCs become detectable.
Gain in sensitivity and cellular detection following labeling with DPPE 19F nanoparticles
In order to reach the goal of this study (to identify the influence of PE nanoparticle enrichment on signal sensitivity in vivo), we next wanted to identify the 19F signal and detection limit within defined regions of interest. For this we selected DC numbers used in the cell dilution curves and performed combined 19F/1H MRI (Figure 6A) as well as voxel-based 19F PRESS in order to quantify the 19F signal within this defined region (Figure 6B). We used the same radio frequency volume coil used for tracking DCs in mice, in order to be able to project our findings to the in vivo experiment. For the volume used to embed cells in agarose (100 μl) within NMR tubes it was sufficient to place a (5 × 5 × 5) mm3 PRESS voxel that covered the entire sample (Figure 6B). The 19F/1H MR imaging experiments showed that 2.5 × 106 was the minimum amount of cells detected in the case of basic-NP labeled DCs and 0.3 × 106 was the minimum for the DPPE-NP labeled DCs (Figure 6A). This corresponds to a cell detection limit of 1.7 × 106 DCs per mm3 for basic-NP labeled DCs and 2 × 105 DCs per mm3 for DPPE-NP labeled DCs for an 19F MR scan time of 16.85 minutes (NEX = 64). The voxel-based spectroscopy method was more sensitive in detecting 19F signal: 0.5 × 106 basic-NP labeled DCs and 0.15 × 106 DPPE-NP labeled DCs could be detected with this method (Figure 6B). This corresponds to a cell detection limit of 4 × 103 DCs per mm3 for basic-NP labeled DCs and 1.2 × 103 DCs per mm3 for DPPE-NP labeled DCs for an 19F MR scan time of 13 minutes (NEX = 512).

19F/1H MRI and Voxel-based 19F MRS of DCs in vitro and in vivo.
(a) 3D rendering of images of PFCE standard (4 × 25 mM) and different number of DCs (labeled with basic or DPPE-enriched nanoparticles) suspended in agarose gel. Images for both 19F and1H nuclei were acquired using the bSSFP sequence and post-processing of data using ImageJ (National Institutes of Health, USA, http://imagej.nih.gov/ij). (b) After acquisition of the bSSFP images, a (5 × 5 × 5) mm3 voxel was placed over the whole region of cells in agarose and spectra were acquired using a PRESS-protocol for 19F MRS (see Methods). (c) DCs (107) labeled with DPPE-enriched nanoparticles were intradermally injected in the left hind limb and DCs (107) labeled with basic nanoparticles in the right hind limb of C57BL/6 mice. Three hours following intradermal application a (3 × 3 × 3) mm3 voxel was placed around left and right popliteal lymph node and the 19F content measured within each lymph node using PRESS sequence as described above.
To study the signal and migration efficiency of DPPE-NP labeled DCs in vivo, we next applied DCs (107) to the left hind limbs of C57BL/6 mice and compared their 19F signal to those labeled with basic 19F nanoparticles (right hind limb). Three hours following intradermal application we placed a (3 × 3 × 3) mm3 PRESS voxel around both left and right popliteal lymph node and measured the 19F signal within each lymph node using PRESS sequence as described above. Both from the 19F/1H MR imaging as well as from the voxel-based spectra illustrating the region-specific 19F signal we observed that DPPE-NP labeled DCs gave a more prominent signal in vivo than the basic-NP labeled DCs (Figure 6C). Using the cell calibration curves described above (Figure 5B) and the FID values at t = 0 for the PRESS spectra, we could translate the quantified 19F signal to the number of cells within the specific lymph node regions: we calculated that after 3 hours 2.85 × 106 DPPE-NP labeled DCs were present in the left lymph node and 0.79 × 105 basic-NP labeled DCs in the right lymph nodes (Figure 6C).
Discussion
Phosphatidylethanolamines (PEs) are aminophospholipids that constitute an integral part of biological membranes. However, PE analogs have also been employed as components of non-biological systems such as non-viral transfection agents21,22 and as coatings of poorly soluble carbon nanotubes to promote cellular uptake23. In the cell, PEs are mainly found in the inner leaflet of the lipid bilayer27 and make up over one fourth of the whole membrane28. PEs play a role in membrane fusion and in cell division29. These phospholipids are translocated to intracellular surfaces via ATP-dependent mechanisms to maintain asymmetrical distribution within the membrane leaflet30,31. Aminophospholipid asymmetry is necessary for preserving the viability of the cell18. The phospholipids PE, PC (phosphatidylcholine), PS (phosphatidylserine), PI (phosphatidylinositol) and SM (sphingomyelin) are irregularly distributed throughout the cellular membrane (23:43:12:9 of PC:PE:PS:PI/SM) in dendritic cells32. The variable distribution of phospholipids is necessary for endocytosis in DCs33,34.
In this study we investigated the influence of two PE analogs on the uptake of fluorine-rich nanoparticles into DCs. We made use of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) to enrich basic nanoparticles with PE. These PEs possess different phase transition temperatures (POPE: +25°C35; DPPE: +64°C36) and their assemblies have very different fluidity. One important difference between the two analogs is the presence of an unsaturated bond within one of the fatty acid hydrocarbon chains in POPE; both fatty acid chains in DPPE are made up of only saturated bonds (Figure 1A).
We found that anchoring DPPE to nanoparticle structures dramatically improved their uptake by DCs (Figures 1–3, Figures 5,6). We also observed an increase in cellular uptake when incorporating POPE into the nanoparticle structure; however the increased uptake was not as remarkable when compared to DPPE (Figure 1B). DSPE-PEG2000 was without any effect (Figures 1–2). In our study we chose PEG2000, with an intermediate molecular weight, in order to stabilize DSPE-based nanoparticles and secure their uptake by DCs. Larger PEG chains are associated with reduced phagocytosis: PEG2000 nanoparticles are taken up more efficiently by macrophages than PEG4000, PEG5000 and PEG10000 coated nanoparticles26,37. Earlier studies reported that covering nanoparticles with higher MW PEG (e.g. PEG5000) prolongs their circulation time in vivo38,39,40, probably due to the decreased phagocytosis and thereby protection from the reticuloendothelial system26,37. In addition to cell uptake and circulation time, the length of the PEG block influences other biological aspects such as target recognition and uptake of delivery systems mediated by address molecules. While long PEG chains may cover the vectors, short PEG chains do not inhibit their enzymatic degradation41. Phospholipid-based drug delivery systems modified with PEG2000 are highly efficient and specific at drug targeting in tumor tissue and remain longer in tumor tissue compared to other DSPE-based carriers with other PEG masses42,43,44. Molecular targeting and cellular uptake can be further enhanced by modification with vector molecules such as receptor-recognizing and cell-penetrating peptides45,46. In our study we did not modify the DSPE-PEG and do not observe any changes in uptake by DCs. Our finding correlates with transfection studies that showed that increasing number of methyl or methylene groups are progressively less active in transfection47. Furthermore, PEG-lipids were found to impair transfection efficacy22. In future it will be interesting to study the impact of peptide-modified DSPE-PEG nanoparticles to promote cellular uptake in DCs46,48.
The observation that DPPE is superior to POPE with regard to cellular uptake is however surprising since liposomal transfection formulations doped with PE analogs revealed that analogs with increasing acyl chain saturation were progressively less active than unsaturated analogs47. In our study we enriched nanoparticles generated from emulsification of PFCE with Pluronic with DPPE. Since PE is strictly bound to the cytosolic leaflet, the observed increased efficiency of internalization of DPPE-enriched nanoparticles by DCs could be explained by an active process translocating PE to the inner membrane surface. It has been shown that multiple unsaturation of the fatty acyl chain significantly decreases the interaction with cholesterol in both bilayers and monolayers19. The success of conveying nanocomplexes into the intracellular compartment is dependent on several factors: hydrophobic alkyl side chains, saturation of C-C bonds in hydrophobic moieties, the head group of the phospholipids utilized to make up the nanocarrier. In a study comparing the combination of a number of phospholipids to liposomal nanoparticles used for gene delivery, combinations containing DPPE and DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) were most effective with regards to cellular transfection49.
PE and PS aminophospholipids are present in cell-derived membrane vesicles: microparticles, microvesicles, exosomes50. Recently a study reported that PE is extensively found on the external surface of microparticles derived from various human cellular sources51. Although cell-derived membrane vesicles were first discovered as a product of platelets during blood coagulation52 and thought to be cellular waste, it is now becoming clear that they play a crucial role in intercellular communication53,54 that may include fusion with their target cells55. Therefore it is conceivable that DPPE-enriched nanoparticles would behave in a similar fashion as these cellular vesicles in order to translocate into intracellular compartments of DCs.
Two clear differences between POPE and DPPE that could explain the differences in cellular uptake are: (i) the presence of an unsaturated bond in POPE and (ii) their dissimilar phase transition temperatures from fluid to liquid crystalline of +25°C for POPE35 and +64°C for DPPE36. These distinct differences between 2 otherwise very similar PEs might determine differences in the fluidity of their assemblies and rigidity of their bilayers, thereby conveying differences in the stability of the particulate structures. The character of the acyl chains in aminophospholipids such as PEs has indeed been shown to be crucial for the fluidity of a monolayer; saturated chains lead to lesser membrane fluidity than unsaturated ones56. It was recently proposed that aminophospholipids with unsaturated oleoyl chains such as POPE are more prominent in areas of protrusion due to their overall conical shape and aminophospholipids consisting of solely saturated palmitoyl chains (that are considered less conical/more cylindrical in shape) do not participate in curvature formation57. Curvature stress is indeed antagonized by addition of more cylindrical lipids58. Changes in the fluidity of biological membranes is also attributed to changes in membrane-initiated signaling processes; for instance, alcohol potentiation of calcium-activated potassium channels is favored by cylindrical phospholipids and blunted by conical ones, regardless of phospholipid head group charge59. Differences in membrane fluidity for DPPE and POPE nanoparticles might influence their interaction with DCs and perhaps the mechanism of entry into the cells. In future, it will be interesting to study the mode of internalization for different rigid and fluidic nanostructures by performing inhibition studies of specific uptake routes (e.g. energy-dependent processes, clathrin-mediated endocytosis, caveolae-mediated endocytosis, macropinocytosis)60 and to follow nanoparticle uptake over time using time-lapse video microscopy61. State-of-the-art technologies such as computational modeling62 or four-dimensional electron microscopy63 will shed light in the future on structure–property relationships of nanocomposites and single-nanoparticle structural dynamics.
In the present study we showed that the incorporation of DPPE in 19F nanoparticles for labeling of DCs increases the intracellular 19F signal in DCs by at least one order of magnitude compared to 19F nanoparticles devoid of DPPE. The observation of an increased uptake of 19F nanoparticles was demonstrated by 19F MR spectroscopy as well as electron and laser scanning microscopy. The microscopy studies demonstrated that nanoparticles are located inside discrete cytosolic endosomes after internalization by DCs, excluding the possibility of cell surface attachment. The agarose spot chemotaxis assay showed that DCs were less capable at moving towards a chemokine concentration when increasing 19F label concentration. Although the migration capability of DCs labeled with DPPE-enriched fluorine nanoparticles is diminished when compared to non-enriched nanoparticles, when given at the same PFCE labeling concentration, lower concentrations of DPPE-enriched fluorine nanoparticles still resulted in a higher 19F signal per million cells and minimal decrease in migration, implying a possible threshold for aminophospholipid enriched nanoparticle labelling concentration that increases the 19F MRS signal without influencing DC migration ability. The order of magnitude increase in 19F signal we observed with 19F spectroscopy could be translated into two advantages: (i) we could decrease the concentration of the 19F label (PFCE) from 20 μmol (employed in our original studies5,6,12) to 5 μmol per 107 DCs and thus decrease the impact of the 19F label on cell migration and (ii) the sensitivity gain considerably improved the cell detection limit to enable imaging and quantification of DCs within specific anatomical regions in vivo.
In conclusion, we present evidence that an enrichment of 19F nanoparticles with DPPE aminophospholipids enhances their uptake by DCs, thereby promoting cellular detection by various 19F MRI and MRS methods. By promoting cellular uptake of 19F label in cellular transplants — such as in DC vaccines for cancer therapy — we contribute to some of the solutions required to overcome the limitations and challenges of cellular MR imaging, particularly with respect to the barriers pertaining to detection limit and signal sensitivity.
Methods
Nanoparticle preparation
Nanoparticles with high fluorine content were prepared by emulsifying Perfluoro-15-crown-5-ether (PFCE, Fluorochem, Derbyshire, UK) via direct sonication, using a ultrasonic homogenizer (Hielscher Ultrasonic GmbH, Teltow, Berlin, Germany). PFCE was emulsified in Pluronic F-68 (Sigma-Aldrich, Germany) for 10 minutes on ice (1.2 M end concentration) to generate a basic formulation: Basic 19F nanoparticles (NP). To enrich the basic particles with DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine, Figure 1A, Avanti Polar Lipid, Inc., Alabaster, AL) diluted Pluronic-basic nanoparticles were mixed with DPPE and further emulsified using the same conditions to obtain a final PFCE concentration of 400 mM and varying DPPE concentrations of 2.5–25 μM (DPPE 19F NP). The same procedure was used to prepare the Pluronic-Rhodamine-DPPE (DPPE-Rhodamine 19F NP) and nanoparticles enriched with POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, Figure 1A, Avanti Polar Lipid, Inc., Alabaster, AL). DSPE-PEG2000—PFCE nanoparticles were prepared by first coating a vial with 10 mg of DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N[methoxy(polyethylene glycol) -2000]), Avanti Polar Lipid, Inc., Alabaster, AL) and then sonicating PFCE in the vial for 10 minutes on ice, to achieve a final concentration of 120 mM PFCE and 3.75 mM DSPE-PEG2000.
Zeta potential and other physical characteristics of nanoparticles
To study the physical characteristics of the above nanoparticles, dynamic light scattering (DLS) data was obtained using a Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK), backscatter detection at 173 degrees and 635 nm laser module at 25°C. DLS provides information on a number of parameters including intensity-weighted z-average diameter, peak diameter and width, zeta (ζ) potential and polydispersity index (PdI). All these physical characteristics of the different nanoparticles were documented and are shown in Table 1. The z-average diameter was used for particle size since it gives an intensity-weighted harmonic diameter and is ideal for comparing different analyses. The PdI is extrapolated from the DLS function and quantitatively describes the particle size distribution best. PdI ranges from 0.01 for monodispersed particles to 0.7 for particles that have a very broad size distribution. The generated nanoparticles have a PdI < 0.3, indicating a relatively low polydispersity and narrow size distribution. The ζ potential was also indirectly determined by measuring the electrophoretic mobility of the nanoparticles under a constant voltage of 40 mV at 25°C.
Cryotransmission Electron Microscopy (Cryo-TEM) of Nanoparticles
Nanoparticle preparations were plunge-frozen onto glow discharged holey carbon grids (Quantifoil Micro Tools, Jena, Germany) in liquid ethane using the environment-controlled Vitrobot (Vitrobot MarkIV, FEI, Eindhoven, The Netherlands). Briefly, 3 μl of the nanoparticle solution were applied onto the grid which was held by tweezers inside the climate chamber (22°C, 100% relative humidity) of the Vitrobot. The solution was automatically blotted with filter paper leaving a thin film of the nanoparticle solution over the holes. The film was allowed to relax for 10 s prior to plunge freezing the sample on the grid in liquid ethane cooled near to its freezing point. Vitrified nanoparticle samples were imaged at −170°C using a Gatan cryo-transfer holder (Gatan 626, Gatan Inc. Pleasanton, USA) and standard low-dose imaging conditions (1 e−/Å2.s)64 at a Tecnai G2 F20 transmission electron microscope (FEI, Oregon, USA), operated at 200 kV. Images were acquired at × 25,000 magnification on a 2k × 2k CCD camera (894 Ultrascan 1000, Gatan Inc., Pleasanton, USA).
DC Preparation
DCs were prepared from bone marrow (BM) suspensions as previously described65. Briefly, BM from femurs of C57BL/6 mice were grown in RPMI-1640 medium containing 10% FCS (Biochrom, Germany) and supplemented with 30 ng/ml GM-CSF. After 9 days in culture, DCs were incubated overnight in the presence of different 19F nanoparticle preparations (end PFCE concentration ranging from 0.1–2 mM or 2–40 μmol/106 cells depending on the experiment) and 1 μg/ml full-length chicken EndoGrade ovalbumin (endotoxin conc.<1 EU/mg; Hyglos, Regensburg, Germany) and 0.5 μg/ml lipopolysaccharide (LPS). Following incubation, unbound 19F nanoparticles were washed thoroughly from the culture dishes by washing with warm PBS. DCs were then harvested and prepared for the ensuing experiments.
Transmission Electron Microscopy (TEM) of DCs
DCs were fixed for 24 hours in PBS containing 2% glutaraldehyde and postfixed for 2 h with 1% osmium tetroxide. Cell pellets were then dehydrated in an ascending series of ethanol and embedded in Poly/Bed 812 (Polysciences, Eppelheim, Germany). Ultrathin sections were stained with uranyl acetate and lead citrate. Sections were imaged using a FEI Morgagni electron microscope (FEI, Eindhoven, The Netherlands) and iTEM software5.
Laser Scanning Microscopy (LSM) of DCs
Intracellular fluorescence in DCs labeled with fluorescently-labeled nanoparticles was investigated using an LSM780 laser scanning microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany). DCs were harvested on day 9, washed and seeded onto 4-chamber μ-slide plates (Ibidi GmbH, München, Germany) and after 4 h labeled with 19F nanoparticles. Prior to LSM, unbound nanoparticles were washed thoroughly from the μ-slide plates by washing with warm culture medium.
Chemotaxis of DCs
DC motility towards chemokine following labeling with 19F nanoparticles was determined using an agarose spot assay, as previously described66. Briefly, a 0.5% agarose solution was prepared, cooled to 40°C and mixed with chemokine (2000 ng/ml CCL21) or PBS (as –ve control); thereafter the agarose solution was pipetted as 10 μl spots onto 35-mm glass dishes (MatTek Corporation, Ashland, MA, USA). Following harvesting, DCs labeled with 19F nanoparticles were introduced, together with culture medium, to the glass dishes and incubated for 4 h at 37°C (5% CO2). The number of cells entering the agarose spot was determined by acquiring and fusing the microscope images for all the fields of views (FOV) making up the 10 μl agarose spot and counting all cells entering the corresponding spot. Image processing (including FOV fusion) and analysis (mainly cell counting) were done with Fiji (Image JA v1.47p, Open source software, NIH, Bethesda, Maryland, USA)67. For fusing all images the Stitching plugin was used and for counting the number of cells within the spot a semi-automatic procedure using the 3D Objects Counter analyze tool was used.
In vitro 19F MR Spectroscopy (MRS)
Following harvesting, DCs (106) labeled with 19F nanoparticles were fixed in 2% PFA and transferred to NMR tubes (external diameter: 4.947 ± 0.019 mm; Wall thickness: 0.043 ± 0.02 mm; VWR International GmbH, Darmstadt, Germany) and the uptake of 19F nanoparticles monitored by 19F spectroscopy. For this we employed an in-house built 19F-tuned loop RF coil5 for signal transmission and reception on a 9.4 T animal MRI scanner (Biospec 94/20 USR, Bruker Biospin, Ettlingen, Germany). A 90° block pulse with 10 kHz bandwidth was used for 19F signal excitation. Spectral representation was done by performing a fast Fourier transformation (FFT) of the acquired free induction decay (FID). The amount of PFCE in each sample was calculated from the amplitude of the extrapolated monoexponential decay, at t = 0 of the FID, which is proportional to the 19F concentration. A standard consisting of 500 mM PFCE was used as quantitative reference in all of these experiments. To determine the cell detection limit of 19F labeling, voxel-based 19F spectroscopy was performed in phantom experiments using increasing numbers of 19F-labeled DCs. Fixed cells were suspended in 2% agarose and transferred to NMR tubes. An NMR tube holder made of acrylonitrile butadiene styrene (ABS) material was printed in-house using a 3D rapid prototyping system (BST 1200es, Dimension Inc., Eden Prairie, MN, USA) and customized to fit within a 1H/ 19F dual-tunable volume RF coil (35 mm inner diameter, 50 mm length; Rapid Biomed, Würzburg, Germany). For quantification of the 19F content within the fixed cells in the NMR tubes, we employed Point REsolved SpectroScopy (PRESS) single voxel spectroscopy. For this purpose we placed a (5 × 5 × 5) mm3 voxel within the region of interest covering the whole cells in agarose (Figure 6). Then we employed the FastMap68 method for volume specific magnetic field (B0) shimming. After B0 shimming, the spectra were acquired using a PRESS-protocol for 19F MRS: TR = 1500 ms, TE = 11.6 ms, voxel size (5 × 5 × 5) mm3, number of repetitions = 512, scan time = 13 min.
In vitro 19F and 1H MRI of Dendritic Cells
To image the fluorine content within the NMR tubes holding the fixed cells labeled with 19F nanoparticle we performed 3D balanced steady state free precession (bSSFP) MRI on the 9.4 T animal MRI using a custom made 1H/19F dual-tunable volume birdcage resonator (Rapid Biomed, Würzburg, Germany) with 35 mm inner diameter and 50 mm length. The bSSFP sequence was chosen because it was also used for the in vivo experiments (described below); it has been shown previously to provide very high SNR, allowing for high spatiotemporal resolution image acquisitions in reasonable scan times and because it produces very good soft tissue contrast, related to their T2/T1 relaxation times69. The scan parameters for bSSFP were as follows for proton (1H) scans: TR = 6.7 ms, TE = 3.3 ms, flip angle = 30°, matrix = 256 × 128 × 128, field of view (FOV) = (5.8 × 2.9 × 5.8) cm3, (227 × 227 × 453) μm3 spatial resolution, number of excitations (NEX) = 1, scan time = 63 sec. For fluorine (19F) scans the parameters for bSSFP were: TR = 3.6 ms, TE = 1.8 ms, flip angle = 30°, matrix = 64 × 32 × 32, FOV = (5.8 × 2.9 × 5.8) cm3, (906 × 906 × 1813) μm3 spatial resolution, NEX = 64, scan time = 16.85 min. To reduce banding artefacts across the SSFP images for both nuclei, 4 acquisitions were made using a 0°, 90°, 180° and 270° phase cycling scheme. The four acquisitions were combined by the sum of squares method using ImageJ (National Institutes of Health, USA, http://imagej.nih.gov/ij).
In vivo 19F 1H MRI and 19F MRS
Animal experiments were carried out in accordance with the guidelines provided and approved by the Animal Welfare Department of the LAGeSo State Office of Health and Social Affairs Berlin (Permit G0070/09: Migration v. Immunzelltherapien). Following harvesting, 19F-labeled DCs were thoroughly washed in serum-free buffer and administered intradermally (5 × 106) into the hind limb of C57BL/6 mice and imaged between 4–18 h following injection. Shortly before and during the MR session, mice were anesthetized using a mixture of isoflurane as inhalation narcosis (0.5–1.5%), pressurized air and oxygen. Mice were imaged on the 9.4 T animal MRI using the same 1H/19F dual-tunable volume birdcage resonator as above (Rapid Biomed, Würzburg, Germany) and the same 3D bSSFP pulse sequences. The temperature of the mice was regulated at 37°C. The respiration rate and temperature was monitored by a remote monitoring system (Model 1025, SA Instruments Inc., NY, USA). For quantification of the 19F content within the lymph node regions, we employed the PRESS sequence as above after placing a (3 × 3 × 3) mm3 voxel around the lymph nodes. After FastMap for volume selective B0 shimming, 19F spectra within the lymph nodes were acquired using the same PRESS-protocol for 19F MRS as above.
MR Data Collection and Analysis
For both in vivo and in vitro MR-measurements a Redhat RHEL4 system and Paravision v5 (Bruker Biospin, Ettlingen, Germany) software were used. NMR data processing, analysis and spectral presentation were performed in the Matlab environment (Matlab version 7.10.0.499, R2010a, The MathWorks, Inc., Natick, Massachusetts, USA). Differences between groups were analyzed by the student t-test. A p-value of p < 0.05 was considered to be statistically significant.
References
Dekaban, G. A. et al. Tracking and evaluation of dendritic cell migration by cellular magnetic resonance imaging. Wiley interdisciplinary reviews. Nanomedicine and nanobiotechnology 5, 469–483, 10.1002/wnan.1227 (2013).
Palucka, K. & Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 39, 38–48, 10.1016/j.immuni.2013.07.004 (2013).
Ahrens, E. T., Flores, R., Xu, H. & Morel, P. A. In vivo imaging platform for tracking immunotherapeutic cells. Nat.Biotechnol. 23, 983–987 (2005).
Srinivas, M., Heerschap, A., Ahrens, E. T., Figdor, C. G. & de Vries, I. J. (19)F MRI for quantitative in vivo cell tracking. Trends Biotechnol. 28, 363–370 (2010).
Waiczies, H. et al. Perfluorocarbon particle size influences magnetic resonance signal and immunological properties of dendritic cells. PLoS One 6, e21981 (2011).
Waiczies, H. et al. Monitoring Dendritic Cell Migration using 19F/1H Magnetic Resonance Imaging. J Vis Exp. 10.3791/50251 (2013).
de Vries, I. J. et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat.Biotechnol. 23, 1407–1413 (2005).
Brooks, R. A., Brunetti, A., Alger, J. R. & Di Chiro, G. On the origin of paramagnetic inhomogeneity effects in blood. Magn Reson.Med. 12, 241–248 (1989).
Lee, N., Inouye, M. & Lauterbur, P. C. 19F- and 13C-NMR studies of a specifically labelled lipoprotein in the Eschericia coli membrane. Biochemical and biophysical research communications 78, 1211–1218 (1977).
Holland, G. N., Bottomley, P. A. & Hinshaw, W. S. F-19 Magnetic-Resonance Imaging. Journal of Magnetic Resonance 28, 133–136 (1977).
Liu, M. S. & Long, D. M. Perfluoroctylbromide as a diagnostic contrast medium in gastroenterography. Radiology 122, 71–76 (1977).
Waiczies, H. et al. Visualizing brain inflammation with a shingled-leg radio-frequency head probe for 19F/1H MRI. Sci.Rep. 3, 1280 (2013).
Bégué, J.-P. & Bonnet-Delpon, D. Bioorganic and medicinal chemistry of fluorine. (John Wiley & Sons, 2008).
Gerebtzoff, G., Li-Blatter, X., Fischer, H., Frentzel, A. & Seelig, A. Halogenation of drugs enhances membrane binding and permeation. Chembiochem : a European journal of chemical biology 5, 676–684, 10.1002/cbic.200400017 (2004).
Smart, B. E. Fluorine substituent effects (on bioactivity). J Fluorine Chem 109, 3–11, Doi 10.1016/S0022-1139(01)00375-X (2001).
Banchereau, J. & Palucka, A. K. Dendritic cells as therapeutic vaccines against cancer. Nature Reviews Immunology 5, 296–306 (2005).
Ahrens, E. T., Helfer, B. M., O'Hanlon, C. F. & Schirda, C. Clinical cell therapy imaging using a perfluorocarbon tracer and fluorine-19 MRI. Magnetic Resonance in Medicine, doi: 10.1002/mrm.25454 (2014).
Balasubramanian, K. & Schroit, A. J. Aminophospholipid asymmetry: A matter of life and death. Annu Rev Physiol 65, 701–734, DOI 10.1146/annurev.physiol.65.092101.142459 (2003).
Smaby, J. M., Brockman, H. L. & Brown, R. E. Cholesterols Interfacial Interactions with Sphingomyelins and Phosphatidylcholines - Hydrocarbon Chain Structure Determines the Magnitude of Condensation. Biochemistry-Us 33, 9135–9142, Doi 10.1021/Bi00197a016 (1994).
Sprong, H., van der Sluijs, P. & van Meer, G. How proteins move lipids and lipids move proteins. Nat Rev Mol Cell Biol 2, 504–513 (2001).
Felgner, J. H. et al. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. Journal of Biological Chemistry 269, 2550–2561 (1994).
Hyvönen, Z. et al. Dioleoyl phosphatidylethanolamine and PEG–lipid conjugates modify DNA delivery mediated by 1, 4-dihydropyridine amphiphiles. Journal of Controlled Release 99, 177–190 (2004).
Antonelli, A. et al. Improved cellular uptake of functionalized single-walled carbon nanotubes. Nanotechnology 21, 425101 (2010).
Caliceti, P. & Veronese, F. M. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Advanced drug delivery reviews 55, 1261–1277 (2003).
Allen, T. M., Hansen, C., Martin, F., Redemann, C. & Yau-Young, A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochimica et biophysica acta 1066, 29–36 (1991).
Fang, C. et al. In vivo tumor targeting of tumor necrosis factor-α-loaded stealth nanoparticles: Effect of MePEG molecular weight and particle size. European Journal of Pharmaceutical Sciences 27, 27–36, 10.1016/j.ejps.2005.08.002 (2006).
Zachowski, A. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. The Biochemical journal 294 ( Pt1), 1–14 (1993).
Virtanen, J. A., Cheng, K. H. & Somerharju, P. Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model. Proceedings of the National Academy of Sciences of the United States of America 95, 4964–4969 (1998).
Emoto, K. et al. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proceedings of the National Academy of Sciences 93, 12867–12872 (1996).
Riekhof, W. R. & Voelker, D. R. Uptake and utilization of lyso-phosphatidylethanolamine by Saccharomyces cerevisiae. Journal of Biological Chemistry 281, 36588–36596 (2006).
Muller, K., Pomorski, T., Muller, P., Zachowski, A. & Herrmann, A. Protein-dependent translocation of aminophospholipids and asymmetric transbilayer distribution of phospholipids in the plasma membrane of ram sperm cells. Biochemistry-Us 33, 9968–9974 (1994).
Laulagnier, K. et al. Mast cell-and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem. J 380, 161–171 (2004).
Steinman, R. M., Mellman, I. S., Muller, W. A. & Cohn, Z. A. Endocytosis and the recycling of plasma membrane. The Journal of cell biology 96, 1–27 (1983).
Steinman, R. M., Brodie, S. E. & Cohn, Z. A. Membrane flow during pinocytosis. A stereologic analysis. The Journal of cell biology 68, 665–687 (1976).
Wang, X. & Quinn, P. J. Cubic phase is induced by cholesterol in the dispersion of 1-palmitoyl-2-oleoyl-phosphatidylethanolamine. Biochimica et Biophysica Acta (BBA)-Biomembranes 1564, 66–72 (2002).
Arouri, A., Dathe, M. & Blume, A. Peptide induced demixing in PG/PE lipid mixtures: a mechanism for the specificity of antimicrobial peptides towards bacterial membranes? Biochimica et Biophysica Acta (BBA)-Biomembranes 1788, 650–659 (2009).
Fontana, G., Licciardi, M., Mansueto, S., Schillaci, D. & Giammona, G. Amoxicillin-loaded polyethylcyanoacrylate nanoparticles: Influence of PEG coating on the particle size, drug release rate and phagocytic uptake. Biomaterials 22, 2857–2865, 10.1016/S0142-9612(01)00030-8 (2001).
Klibanov, A. L., Maruyama, K., Torchilin, V. P. & Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett 268, 235–237 (1990).
Klibanov, A. L., Maruyama, K., Beckerleg, A. M., Torchilin, V. P. & Huang, L. Activity of amphipathic poly(ethylene glycol) 5000 to prolong the circulation time of liposomes depends on the liposome size and is unfavorable for immunoliposome binding to target. Biochimica et Biophysica Acta (BBA) - Biomembranes 1062, 142–148, 10.1016/0005-2736(91)90385-L (1991).
Mori, A., Klibanov, A. L., Torchilin, V. P. & Huang, L. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett 284, 263–266, 10.1016/0014-5793(91)80699-4 (1991).
Koren, E., Apte, A., Sawant, R. R., Grunwald, J. & Torchilin, V. P. Cell-penetrating TAT peptide in drug delivery systems: Proteolytic stability requirements. Drug delivery 18, 377–384, 10.3109/10717544.2011.567310 (2011).
Lukyanov, A., Gao, Z., Mazzola, L. & Torchilin, V. Polyethylene Glycol-Diacyllipid Micelles Demonstrate Increased Accumulation in Subcutaneous Tumors in Mice. Pharm Res 19, 1424–1429, 10.1023/a:1020488012264 (2002).
Torchilin, V. P. Micellar Nanocarriers: Pharmaceutical Perspectives. Pharm Res 24, 1–16, 10.1007/s11095-006-9132-0 (2007).
Kale, A. A. & Torchilin, V. P. “Smart” Drug Carriers: PEGylated TATp-Modified pH-Sensitive Liposomes. Journal of Liposome Research 17, 197–203, 10.1080/08982100701525035 (2007).
Sydow, K., Torchilin, V. P. & Dathe, M. Lipopeptide-modified PEG-PE-based pharmaceutical nanocarriers for enhanced uptake in blood–brain barrier cells and improved cytotoxicity against glioma cells. European Journal of Lipid Science and Technology 116, 1174–1183, 10.1002/ejlt.201300373 (2014).
Sauer, I., Dunay, I. R., Weisgraber, K., Bienert, M. & Dathe, M. An apolipoprotein E-derived peptide mediates uptake of sterically stabilized liposomes into brain capillary endothelial cells. Biochemistry-Us 44, 2021–2029 (2005).
Felgner, J. H. et al. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. Journal of Biological Chemistry 269, 2550–2561 (1994).
Sydow, K., Torchilin, V. P. & Dathe, M. Lipopeptide-modified PEG-PE-based pharmaceutical nanocarriers for enhanced uptake in blood-brain barrier cells and improved cytotoxicity against glioma cells. European Journal of Lipid Science and Technology 116, 1174–1183, 10.1002/ejlt.201300373 (2014).
Ramezani, M., Khoshhamdam, M., Dehshahri, A. & Malaekeh-Nikouei, B. The influence of size, lipid composition and bilayer fluidity of cationic liposomes on the transfection efficiency of nanolipoplexes. Colloid Surface B 72, 1–5, DOI 10.1016/j.colsurfb.2009.03.018 (2009).
van Dommelen, S. M. et al. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. Journal of Controlled Release 161, 635–644, DOI 10.1016/j.jconrel.2011.11.021 (2012).
Larson, M. C., Woodliff, J. E., Hillery, C. A., Kearl, T. J. & Zhao, M. Phosphatidylethanolamine is externalized at the surface of microparticles. Biochimica et biophysica acta 1821, 1501–1507, 10.1016/j.bbalip.2012.08.017 (2012).
Wolf, P. The nature and significance of platelet products in human plasma. British journal of haematology 13, 269–288 (1967).
Roseblade, A. et al. Cell-derived microparticles: new targets in the therapeutic management of disease. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques 16, 238–253 (2013).
Hargett, L. A. & Bauer, N. N. On the origin of microparticles: From "platelet dust" to mediators of intercellular communication. Pulmonary circulation 3, 329–340, 10.4103/2045-8932.114760 (2013).
Deregibus, M. C. et al. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 110, 2440–2448, 10.1182/blood-2007-03-078709 (2007).
Saulnier, P., Foussard, F., Boury, F. & Proust, J. Structural properties of asymmetric mixed-chain phosphatidylethanolamine films. Journal of colloid and interface science 218, 40–46 (1999).
Malcharek, S., Hinz, A., Hilterhaus, L. & Galla, H.-J. Multilayer structures in lipid monolayer films containing surfactant protein C: effects of cholesterol and POPE. Biophysical Journal 88, 2638–2649 (2005).
Epand, R. M. & Bottega, R. Determination of the phase behaviour of phosphatidylethanolamine admixed with other lipids and the effects of calcium chloride: implications for protein kinase C regulation. Biochimica et Biophysica Acta (BBA) - Biomembranes 944, 144–154, 10.1016/0005-2736(88)90427-0 (1988).
Crowley, J. J., Treistman, S. N. & Dopico, A. M. Distinct Structural Features of Phospholipids Differentially Determine Ethanol Sensitivity and Basal Function of BK Channels. Mol Pharmacol 68, 4–10, 10.1124/mol.105.012971 (2005).
Gratton, S. E. A. et al. The effect of particle design on cellular internalization pathways. Proceedings of the National Academy of Sciences 105, 11613–11618, 10.1073/pnas.0801763105 (2008).
Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proceedings of the National Academy of Sciences of the United States of America 103, 4930–4934, 10.1073/pnas.0600997103 (2006).
Yan, L.-T. & Xie, X.-M. Computational modeling and simulation of nanoparticle self-assembly in polymeric systems: Structures, properties and external field effects. Progress in Polymer Science 38, 369–405 (2013).
van der Veen, R. M., Kwon, O.-H., Tissot, A., Hauser, A. & Zewail, A. H. Single-nanoparticle phase transitions visualized by four-dimensional electron microscopy. Nature chemistry 5, 395–402 (2013).
Adrian, M., Dubochet, J., Lepault, J. & McDowall, A. W. Cryo-electron microscopy of viruses. Nature 308, 32–36 (1984).
Bendix, I. et al. MAPK3 deficiency drives autoimmunity via DC arming. European journal of immunology 40, 1486–1495, 10.1002/eji.200939930 (2010).
Wiggins, H. & Rappoport, J. An agarose spot assay for chemotactic invasion. BioTechniques 48, 121–124, 10.2144/000113353 (2010).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, Doi 10.1038/Nmeth.2019 (2012).
Gruetter, R. Automatic, localized in vivo adjustment of all first- and second-order shim coils. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine/Society of Magnetic Resonance in Medicine 29, 804–811 (1993).
Economopoulos, V., Noad, J. C., Krishnamoorthy, S., Rutt, B. K. & Foster, P. J. Comparing the MRI appearance of the lymph nodes and spleen in wild-type and immuno-deficient mouse strains. PLoS One 6, e27508, 10.1371/journal.pone.0027508 (2011).
Acknowledgements
This study was funded by the Deutsche Forschungsgemeinschaft to SW (DFG WA 2804). We thank Ms. Stefanie Kox and Ms. Yvonne Balke for excellent technical support.
Author information
Authors and Affiliations
Contributions
S.W., S.L. and T.N. contributed to the manuscript text; S.W., S.L., K.S., S.D., M.C.K., C.M., D.L., I.S. and B.F. performed the experiments; S.W., S.L., H.R.M., H.W., M.D. and A.P. analyzed the data. All authors reviewed the manuscript.
Ethics declarations
Competing interests
S.W. received research grants and poster honoraria from Novartis. K.S. was partly financed by the DFG (DA 324/9-1). H.W. is employed by and T.N. is founder of MRI.TOOLS GmbH. T.N. received speaker honoraria from Siemens Healthcare, Erlangen, Germany. M.D. now works with Siemens Healthcare. S.L., S.D., M.C.K., C.M., D.L., I.S., H.R.M., B.F., M.D. and A.P. have nothing to disclose.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Waiczies, S., Lepore, S., Sydow, K. et al. Anchoring Dipalmitoyl Phosphoethanolamine to Nanoparticles Boosts Cellular Uptake and Fluorine-19 Magnetic Resonance Signal. Sci Rep 5, 8427 (2015). https://doi.org/10.1038/srep08427
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08427
This article is cited by
-
Toward 19F magnetic resonance thermometry: spin–lattice and spin–spin-relaxation times and temperature dependence of fluorinated drugs at 9.4 T
Magnetic Resonance Materials in Physics, Biology and Medicine (2019)
-
Fluorine-19 MRI at 21.1 T: enhanced spin–lattice relaxation of perfluoro-15-crown-5-ether and sensitivity as demonstrated in ex vivo murine neuroinflammation
Magnetic Resonance Materials in Physics, Biology and Medicine (2019)
-
Enhanced Fluorine-19 MRI Sensitivity using a Cryogenic Radiofrequency Probe: Technical Developments and Ex Vivo Demonstration in a Mouse Model of Neuroinflammation
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
