
Abstract
Understanding the kinetic implication of solid-solution vs. biphasic reaction pathways is critical for the development of advanced intercalation electrode materials. Yet this has been a long-standing challenge in materials science due to the elusive metastable nature of solid solution phases. The present study reports the synthesis, isolation and characterization of room-temperature LixMn1.5Ni0.5O4 solid solutions. In situ XRD studies performed on pristine and chemically-delithiated, micron-sized single crystals reveal the thermal behavior of LixMn1.5Ni0.5O4 (0 ≤ x ≤ 1) cathode material consisting of three cubic phases: LiMn1.5Ni0.5O4 (Phase I), Li0.5Mn1.5Ni0.5O4 (Phase II) and Mn1.5Ni0.5O4 (Phase III). A phase diagram capturing the structural changes as functions of both temperature and Li content was established. The work not only demonstrates the possibility of synthesizing alternative electrode materials that are metastable in nature, but also enables in-depth evaluation on the physical, electrochemical and kinetic properties of transient intermediate phases and their role in battery electrode performance.
Similar content being viewed by others

Introduction
A significant number of Li-ion batteries (LIBs) positive electrode materials operate in a first-order phase transition mechanism where the equilibrium electrochemical potential (E) of the electrode does not vary with the ratio of the phases present in the system1,2,3,4. Two-phase transformation has traditionally been associated with severe limitations, including slow kinetics and poor stability of the materials. The large volume change between the involved phases leads to the accumulation of strain and mechanical degradation of the active particles and the creation of Li ions transport barriers at the phase boundaries5. Various approaches, including downsizing the particles6 and creating anti-site defects in the lattice7, were proposed to reduce the miscibility gap and promote single-phase transformation in LiFePO4. A recent report describes the use of in situ XRD with high temporal resolution to capture the appearance of transient solid solutions during high-rate cycling of LiFePO4 nanoparticle electrodes8. The formation of LixFePO4 (0 < x < 1) solid solutions was also accomplished by heating various mixtures of LiFePO4 and FePO4 to about 350°C9,10. However, room-temperature single phases needed for detailed characterization were not attainable due to their high metastability.
LiMn1.5Ni0.5O4 (LMNO) is a promising positive electrode material for high-energy density LIBs due to its 3D lithium-ion diffusion paths, a high operating voltage of 4.7 V and a theoretical capacity of 147 mAh/g (energy density of 700 Wh/kg)11,12,13,14. The spinel transforms through two topotactic two-phase reactions involving three cubic phases: LiMn1.5Ni0.5O4 is first oxidized to  and then
and then  during charge and vice versa during discharge15,16. Here
during charge and vice versa during discharge15,16. Here  denotes the vacant octahedral sites in the structure. The volume changes of Phase I/II and Phase II/III are approximately 3% and 3.3%, respectively. Several parameters, including particle surface facets17, particle size18,19,20, substitution16,21,22 and structural ordering11,23,24, have been shown to have a large influence on the phase transformation pathway. First-principles calculation suggested that in perfectly disordered LMNO, single phase transformation throughout the entire Li composition range is possible at room temperature25. Experimental studies, on the other hand, reported the presence of a large solid solution region followed by one or two two-phase transformations in disordered LMNO17,26. For ordered LMNO, two two-phase transitions between three cubic phases were typically observed, with the Li content range of Phase I solid solution varying significantly among reports27,28. Although single-phase transformation is considered kinetically advantageous over the two-phase process, owing to the observation that disordered LMNO with a larger solid solution region is often associated with better performance29, the kinetic role of solid solution is largely unknown.
denotes the vacant octahedral sites in the structure. The volume changes of Phase I/II and Phase II/III are approximately 3% and 3.3%, respectively. Several parameters, including particle surface facets17, particle size18,19,20, substitution16,21,22 and structural ordering11,23,24, have been shown to have a large influence on the phase transformation pathway. First-principles calculation suggested that in perfectly disordered LMNO, single phase transformation throughout the entire Li composition range is possible at room temperature25. Experimental studies, on the other hand, reported the presence of a large solid solution region followed by one or two two-phase transformations in disordered LMNO17,26. For ordered LMNO, two two-phase transitions between three cubic phases were typically observed, with the Li content range of Phase I solid solution varying significantly among reports27,28. Although single-phase transformation is considered kinetically advantageous over the two-phase process, owing to the observation that disordered LMNO with a larger solid solution region is often associated with better performance29, the kinetic role of solid solution is largely unknown.
Encouraged by previous studies on temperature-dependent solid solution behavior in the LiFePO4/FePO4 system, we performed comprehensive thermal studies on a series of chemically delithiated LixMn1.5Ni0.5O4 (LixMNO, 0 ≤ x < 1) prepared from structurally ordered LMNO crystals with exclusive (111) surface facets. The thermal treatment resulted in a reduction in the miscibility gap between the cubic phases I and II and a series of single-phase solid solutions were attainable even at room temperature. Combining various techniques such as in situ temperature-controlled powder XRD (TXRD), Raman, Fourier transform infrared (FTIR) and X-ray absorption (XAS) spectroscopies, a phase diagram capturing the changes as functions of both temperature and Li content was established and the physical properties of LixMNO solid solutions were investigated.
Results
In situ XRD studies on the thermal behavior of LixMNO (0 ≤ x ≤ 1)
Uniform, micron-sized and octahedron-shaped LiMn1.5Ni0.5O4 crystals were synthesized by a molten salt method, as reported in our previous publication17. Extensive characterization by high-resolution transmission electron microscopy (HRTEM), selected area electron diffraction (SAED), FTIR, nuclear magnetic resonance (NMR) and electrochemical techniques revealed that the sample adopted near perfectly-ordered structure with a space group of P4332 and the particles are enclosed by 100% of (111) surface facets. A series of delithiated LixMNO, with x = 0.90, 0.82, 0.71, 0.51, 0.40, 0.25, 0.11, 0.06 and 0, were prepared by chemical oxidation with varying amounts of a 0.1 M solution of nitronium tetrafluoroborate (NO2BF4) in acetonitrile. The lithium content x, solely controlled by the ratio between the spinel and the oxidizing agent, was determined by ICP analysis. The reactions resulted in a variety of mixtures among three cubic phases: LiMn1.5Ni0.5O4 (Phase I), Li0.5Ni0.5Mn1.5O4 (Phase II) and Ni0.5Mn1.5O4 (Phase III). The XRD patterns and the weight fraction of the phases in the samples were obtained through full-pattern Rietveld refinements and the results are shown in Fig. 1a and 1b, respectively. Low level of lithium extraction (x > 0.71) led to the formation of cubic Phase II at the expense of Phase I. Phase III appeared on further Li removal, which rendered the coexistence of three cubic phases in the samples with an intermediate Li content of 0.25 < x ≤ 0.71, although only about 3% of Phase III was present in Li0.71MNO. At low Li content (x ≤ 0.25), the samples were composed of Phase II and III only. The lattice parameters of the Phases I, II and III were refined to be 8.1687(2), 8.0910(6) and 8.0005(3) Å, respectively. The smaller lattice dimensions in Phase II and III are directly related to the reduced Li content as well as a higher average Ni oxidation state in the spinel lattice.

(a) Room-temperature XRD patterns and (b) phase composition of the as-prepared LixMn1.5Ni0.5O4 crystal samples. Global Li content as indicated which has a standard deviation of less than 0.8%.
It is worth noting that previous in situ XRD studies on the ordered LMNO octahedrons have revealed the existence of solid solution at lower state of charge, to an estimated Li content of 0.59 < x < 117. In the literature, the reported in situ experiments so far showed significant variation in the lithium content range where solid solution behavior was observed. Aside from the differences in the step size used in the individual experiments, subtle variation in sample crystal structure is believed to play a major role17,26,30. Our effort to isolate the single phases from the electrochemically charged electrodes, however, was unsuccessful. After removing the same amount of Li in the x > 0.71 region, nearly identical phase mixtures were obtained from both electrochemically and chemically prepared LixMNO, as confirmed by ex situ XRD patterns collected on the cathodes recovered from partially charged cells (Supplementary Figure 1). This suggests that the solid solutions appearing during electrochemical charge and discharge are metastable transient intermediates that bear kinetic significance but thermodynamically unstable. Currently, no viable approach for the synthesis of phase-pure LixMNO intermediates has been reported.
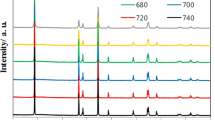
In situ temperature-controlled XRD studies were carried out to systematically investigate the thermal behavior of LixMNO (0 ≤ x ≤ 1). Chemically delithiated samples were used due to its simplicity in preparing large quantities needed for this study as well as the purity in the absence of binder and carbon additives. Fig. 2 shows the TXRD patterns of selected LixMNO compositions (x = 0.82, 0.51, 0.25 and 0) collected during both heating and cooling. For comparison, the patterns of pristine LMNO are also shown in the supplemental information (Supplementary Figure 2), which displayed straightforward thermal expansion and contraction behavior resulting from heating and cooling, respectively. In all cases, no significant changes were detected below 150°C. Between 150 and 225°C, the peaks from the initial cubic phases broadened and started to merge in the samples with x = 0.82 (Fig. 2a) and 0.51 (Fig. 2b) and the complete transformation into a single cubic phase was observed at 250°C. The single phase formation temperature increases along with the decrease of Li content. The reduction in the miscibility gap at the high temperature was attributed to the increase in Li ion diffusion from the lithium-rich to the lithium-poor phase, similar to the phenomena observed in the LiFePO4/FePO4 system. The lowered formation temperature of LixMNO is consistent with the fact that Li mobility is generally higher in LMNO than the LiFePO4 system. The cubic framework of the thermal-driven solid solution maintained during the cooling, with no phase separation observed even at room temperature. In the fully delithiated sample composed of Phase III only (x = 0, Fig. 2d), a new spinel-type phase with expanded lattice dimensions appeared above 200°C, suggesting that Phase III is susceptible to thermal-induced phase conversion. Here, the use of “a new spinel-type phase” is to denote the decomposition products, although it is possible that this phase consists of several structurally similar phases and/or compounds. At 250°C, the sample is composed of the decomposition products and Phase III in an approximately 1:1 weight ratio. Further conversion occurred during the early stage of cooling which led to much larger presence of the spinel-type phase in the cooled MNO at room temperature. For x = 0.25 consisting of Phase II and III in the as-prepared sample, three components, Phase II, the spinel-type phase and Phase III, were observed at 200°C (Fig. 2c). Upon further heating to 250°C, Phase II disappeared and the mixture was composed of the spinel-type phase and Phase III only. Fig. 3 compares the TXRD patterns of the LixMNO series (x = 1, 0.90, 0.82, 0.71, 0.51, 0.40, 0.25, 0.11, 0.06 and 0) collect at 250°C (Fig. 3a) and after cooling to RT (Fig. 3b). At 250°C, single phases were obtained in samples with x ≥ 0.51 which maintained phase pure after cooling to RT. Samples with Li content below 0.51, however, were composed of mixtures at both 250°C and RT. The cooled samples with x ≤ 0.25 consisted mostly of the spinel-type phase which has significantly broader peaks compared to the initial spinel phases in the as-prepared samples.

Temperature-controlled XRD patterns of selected LixMn1.5Ni0.5O4 crystal samples: (a) x = 0.82, (b) x = 0.51, (c) x = 0.25 and (d) x = 0.

XRD patterns of the LixMn1.5Ni0.5O4 crystal samples collected at (a) 250°C and (b) after cooling to 25°C.
The relationship between the lattice parameters of the phases present and the heating temperature is shown in Fig. 4, established from the full-pattern Rietveld refinements of the TXRD patterns. For samples with 0.51 ≤ x < 1 that have minimal presence of Phase III, the cubic lattice parameters grew closer to each other when the temperature is raised above 150°C which eventually became a single lattice parameter around 250°C (Fig. 4a), confirming the formation of a single-phase solid solution in these samples. All solid solutions can be indexed using the same space group of  , with the refinements produced similar lattice parameters in the range of 8.1873(9) to 8.1907(8) Å. This is consistent with the previous observation that in ordered LMNO, the lattice dimension is relatively insensitive to lithium content at high x17. Upon cooling, the changes in lattice parameters followed a near linear relationship as a response to thermal contraction, reaching the range of 8.1687(8) to 8.1720(2) Å (Fig. 4b). The results confirm the absence of structural decomposition or phase demixing during the cooling process, further suggesting that, compared to the much discussed LixFePO4 solid solutions, thermal-driven LixMNO solid solution phases possess higher stability. For samples with 0 < x ≤ 0.40 that have a predominant presence of Phase III (≥22 wt%), the merge of all three lattice parameters was not observed during heating (Fig. 4c). At 250°C, the lattice parameters of Phase II merged into the new spinel-type phase at about 8.1961(4) Å, but Phase III remained in all samples.
, with the refinements produced similar lattice parameters in the range of 8.1873(9) to 8.1907(8) Å. This is consistent with the previous observation that in ordered LMNO, the lattice dimension is relatively insensitive to lithium content at high x17. Upon cooling, the changes in lattice parameters followed a near linear relationship as a response to thermal contraction, reaching the range of 8.1687(8) to 8.1720(2) Å (Fig. 4b). The results confirm the absence of structural decomposition or phase demixing during the cooling process, further suggesting that, compared to the much discussed LixFePO4 solid solutions, thermal-driven LixMNO solid solution phases possess higher stability. For samples with 0 < x ≤ 0.40 that have a predominant presence of Phase III (≥22 wt%), the merge of all three lattice parameters was not observed during heating (Fig. 4c). At 250°C, the lattice parameters of Phase II merged into the new spinel-type phase at about 8.1961(4) Å, but Phase III remained in all samples.

(a) Changes in the lattice parameters upon heating the LixMn1.5Ni0.5O4 crystal samples (0.51 ≤ x ≤ 1), (b) the change in solid-solution lattice parameter on cooling, (c) changes in the lattice parameters upon heating the LixMn1.5Ni0.5O4 crystal samples (0 ≤ x ≤ 0.40) and (d) refined domain size of the main phase (as indicated) in LixMNO at 250°C. Error range in the lattice parameters is less than ±0.001. Phase I: square; Phase II: circle; Phase III: triangle; Spinel-type phase: star.
One possible compound represented by the new spinel-type phase is NiMn2O4-type spinel, which was previously reported as a common thermal decomposition product in Ni and Mn containing ternary oxides. Consistent with our results, Hu et al. have shown that fully delithiated and structurally ordered MNO was thermally unstable which released oxygen and transformed into a NiMn2O4-type phase at a temperature as low as 250°C31. The transition occurs when the tetragonal Li is fully or partially replaced by Mn which renders a much large lattice parameter at room-temperature, up to 8.40 Å with full replacement. Our effort to obtain room-temperature lattice parameter of the newly formed spinel-type phase was not successful as cooling of the samples with x ≤ 0.25 led to significant peak broadening (Supplementary Figure 3) and inaccurate refinement of the XRD patterns.
Fig. 4d compares the refined domain size of the main phase in LixMNO at 250°C, namely the solid solutions in the 0.40 < x ≤ 1 group and the new spinel-type phase in the 0 ≤ x < 0.40 group. The domains size of ~50 nm in the former was reduced to half (~25 nm) in the latter case, consistent with major structural rearrangement and phase transformation associated with the thermal instability of Phase III in the Li-poor samples.
Vibrational spectroscopy of LixMNO (0 ≤ x ≤ 1)
Raman and FTIR spectroscopies probe the vibration modes of the bonds in a single molecule which are powerful in elucidating crystal symmetry of oxide samples. Unlike the powder XRD measurements, both techniques are able to detect small amounts of impurities as well as short-range ordering in amorphous phases that are typically transparent to XRD26,32. Fig. 5a shows the Raman spectra of as-prepared LxMNO samples (left) and their heat-treated counterparts that were recovered from the TXRD studies (right). In the as-prepared samples, typical Raman bands associated with cubic spinel lattice vibrations are displayed in the 400–700 cm−1 region24. The peaks at 404 and 496 cm−1 on the pristine LMNO spectrum are commonly attributed to Eg and F2g(2) Ni2+-O stretching modes. With decreasing x, the disappearance of the peaks at 404 cm−1 and the appearance of the peaks around 485 and 545 cm−1 indicate oxidation of Ni2+ to Ni4+33. The peaks between 570 and 650 cm−1 are assigned as A1g and F2g(1) Mn-O modes in the MnO6 octahedron, with their position and relative intensity ratio closely related to Mn-O bond length, MnO6 distortion and Mn oxidation state. It has been shown that an increase in Mn3+ content, often associated with a loss of translational symmetry due to the Jahn teller effect, leads to the broadening of the bands from both A1g and F2g(1) modes. This broadening may be further enhanced by an increase in electrical conductivity induced by charge transfer via electron hopping between Mn3+ and Mn4+. On the pristine LMNO spectrum, the well-resolved split of the F2g(1) bands at 593 and 612 cm−1, which mainly originate from the vibration modes of the Mn4+-O bonds, is consistent with the ordering of the transition-metal cations (Ni2+ and Mn4+) where supplementary Raman modes are expected to be active. The A1g band at 635 cm−1 is related to both Mn4+-O and Mn3+-O vibrations. The intensity ratio of 593/635, therefore, is traditionally used as an indicator for Mn4+ concentration in the lattice as well as the average Mn oxidation state in the sample34. The dominance of the peak at 593 cm−1 and its continuous shift towards lower frequency along with decreasing lithium content can be attributed to a combination of slight increase in Mn4+/Mn3+ ratio and a decrease in Li ion concentration at 8a sites. As determined by the electrochemical studies in our previous report, the estimated Mn3+ concentration in the pristine octahedron sample is close to 1.5%17.

(a) Raman and (b) FTIR spectra of the LixMNO samples collected at room temperature. Left: as-prepared and right: heat-treated.
No significant differences were observed when comparing the spectra of the as-prepared and heat-treated samples with 0.71 ≤ x ≤ 1, all of which resembled that of the LMNO pristine sample. This further confirms the retention of spinel local symmetry in the thermally-induced solid solutions and phase purity of the samples. On the other hand, substantial peak broadening and reduced scattering intensities were observed in the heated-treated samples with 0 ≤ x ≤ 0.51, suggesting thermal induced structural changes, formation of secondary phases with reduced domain size and possibly, enhanced electrical conductivity35. The dominance of the broad bands at 486 and 588 cm−1 on the spectra suggests enhanced distortion of MnO6 octahedron and the presence of a new spinel phase with reduced symmetry, such as λ-MnO2 or NiMn2O4-type spinel36,37.
The corresponding FTIR spectra of the LixMNO samples before and after heat treatment are shown in Fig. 5b. The presence of the eight well-defined bands at 430, 468, 480, 501, 557, 594, 621 and 650 cm−1 is consistent with the ordered structure in the pristine LMNO sample. Similar to the XRD and Raman results, the cubic spinel structure in the pristine sample was retained and no significant change in symmetry resulted from either delithiation or heat treatment of the samples with 0.71 ≤ x ≤ 1. In the thermally-treated samples, peak broadening and changes in intensity ratios between the bands were first observed in L0.51MNO, which exacerbated as x decreased. Since Li0.51MNO is the first sample in the series where the presence of Phase III becomes significant enough (13 wt% in x = 0.51 as opposed to <3 wt% in x = 0.71, the previous sample in the series) to enable the detection of its decomposition products by the vibrational techniques, the results provide further evidence that the formation of the new spinel-type phase in LixMNO (0 ≤ x ≤ 0.51) is directly related to the thermal decomposition of Phase III. The higher sensitivity of the vibrational spectroscopy in detecting phases in small amounts and with short-range ordering complements the results from the bulk XRD study where the technique limits the detection of impurities in x = 0.51.
X-ray absorption spectroscopy of LixMNO (0 ≤ x ≤ 1)
Hard X-ray absorption spectroscopy correlates the changes in the absorption edge energy to the variations in the average oxidation state of the absorbing atoms being analyzed in the bulk material38,39. The technique also probes the local and electronic structures near the absorbing atoms and it has the ability to reveal element-specific structural changes in the samples. Fig. 6 shows the Ni K-edge X-ray absorption near edge structure (XANES) spectra from the as-prepared (Fig. 6a) and heat-treated (Fig. 6b) LixMNO series, respectively. In the as-prepared samples, chemical oxidation led to a monotonous edge-shift to higher energy, consistent with a continuous increase in average oxidation state of nickel as it transformed from the divalent state in LiMn1.5Ni0.5O4 to tetravalent state in Mn1.5Ni0.5O4. Two isosbestic points, indicative of a three-component system, were observed at 8353.8 and 8355 eV on the XANES spectra, which is consistent with the presence of Phase I, II and III in the as-prepared samples. The results are also in good agreement with the in situ XANES data recently reported by Arai et al.40 For comparison, the room-temperature spectra of the LixMNO samples subjected to the heat treatment are shown in Fig. 6b. Negligible changes were observed in samples with high Li content of 0.71 ≤ x ≤ 1. The samples with lower lithium content of 0 ≤ x ≤ 0.51, on the other hand, experienced substantial edge shift towards lower energy, with the absorption edges nearly overlapping with those of lithium-rich samples (0.71 ≤ x ≤ 1). Fig. 6c compares the edge position, defined by Photoelectron Energy Origin (E0) that is a commonly used parameter for extracting edge energy level in XANES spectra31,39, as a function of lithium content in the samples before and after thermal treatment. Clearly, heating led to significant reduction in Ni oxidation state in the samples with x < 0.71, suggesting that the formation of the new spinel-type phase is related to Ni reduction. Fig. 7 shows the Mn K-edge XANES spectra from the as-prepared and heat-treated samples. Only slight energy increase resulted from chemical oxidation (Fig. 7a), corresponding to the low concentration of Mn3+ in the pristine sample (1.5%) and limited Mn3+/Mn4+ redox activity. Unlike the Ni case, heating led to negligible edge shift in the entire series (Fig. 7b), suggesting the absence of significant Mn redox activities during thermal decomposition of Phase III. The edge position as a function of lithium content before and after the heat treatment is compared in Fig. 7c.

XANES spectra of Ni K-edge: (a) as-prepared, (b) heat-treated and (c) the relationship between the Ni energy edge at the first inflection point (E0) and Li content in the samples.

XANES spectra of Mn K-edge: (a) as-prepared, (b) heat-treated and (c) the relationship between the Ni energy edge at the first inflection point (E0) and Li content in the samples.
Further changes were observed in the intensity of Mn pre-edge absorption peaks arising from the dipole forbidden 1s → 3d electronic transitions, which are typically week in the transition metals. In the presence of 3d and 4p orbital hybridization, often resulting from structural distortion in local symmetry or non-centrosymmetric environment between the metal and oxygen coordination, the transitions are made partially allowed and the peaks become much stronger41,42,43. Also, in an event where a transition metal occupies the tetrahedral instead of the octahedral site, much stronger pre-edge peaks are observed owing to the lack of inversion center in tetrahedral geometry. As shown in Fig. 8, heat-treatment led to negligible changes in samples with 0.71 ≤ x ≤ 1 (Fig. 8a). The treated samples with low lithium contents, particularly those with 0 ≤ x ≤ 0.25, on the other hand, have noticeably increased Mn pre-edge absorption peaks (Fig. 8b). This suggests that although no significant Mn redox activities are involved in the thermal decomposition of Phase III, some Mn cations may migrate from the octahedral to the tetrahedral sites. This is consistent with the possible formation of NiMn2O4-type phase through partial replacement of the tetragonal Li by Mn. Further analysis is necessary in order to determine the chemical nature of the thermal decomposition products.

XANES spectra of Mn pre-edge: (a) 0.71 ≤ x ≤ 1 and (b) 0 ≤ x ≤ 0.25.
Dotted lines: as-prepared and solid lines: heated-treated samples.
Phase diagram of LixMNO (0 ≤ x ≤ 1)
On the basis of the results obtained from TXRD, Raman, FTIR and X-ray absorption spectroscopies, the phase diagram of LixMNO on heating was constructed (Fig. 9). Because of the nature of chemical delithiation, the Li content step size in this study is relatively coarse which limits the resolution of our phase diagram. Overall, the phase behavior of LixMNO as a function of Li content can be divided into three regions at room temperature. In the first region where Li content is between 0.71 and 1, the initial cubic phases I and II merge at high temperature and phase-pure solid solutions form when the temperature is raised above 200°C. The formation temperature increases along with the decrease of Li content. This is the only domain where single phases are attainable in the LixMNO system at both elevated and room temperatures. In the second domain where samples are composed of Phase I, II and III (0.25 < x < 0.71), the merge of Phase I and II led to the formation of solid solution but Phase III remains and a new spinel-type impurity phase also appears at high temperature. Further increasing temperature to above 265°C led to the complete conversion of Phase III to the spinel-type phase (Supplementary Figure 4). For 0 < x ≤ 0.25, no solid solution formation was observed during the heating process. Phase III decomposed to the spinel-type phase and the final treated samples are composed of both phases.

Phase diagram of LixMn1.5Ni0.5O4 on heating.
× indicates the temperature where significant structural changes were observed in the LixMn1.5Ni0.5O4 TXRD patterns. The phase compositions in each region as labeled.
While Phase I and II in LMNO system are able to merge into a single solid solution at high temperature, Phase III has very low solubility in the lithium containing spinel Phase I and/or II. Phase III also has the lowest thermal stability among the three cubic phases, which decomposes into a spinel-type phase upon heating at a temperature as low as 150°C. The decomposition temperature is largely influenced by the presence of Phase I, which decreases along with the reduction in Phase I content in the sample. It appears that in the absence of Phase I, Phase II undergoes thermal decomposition as well but at a temperature much higher than that of Phase III.
Discussion
The study sheds light on the thermal behavior of the LixMNO system and reports the isolation of phase pure room-temperature LixMNO solid solutions for the first time. A series of delithiated LixMNO samples were prepared by chemical delithiation. While the samples with a Li content between 0.71 and 1 were composed of Phase I and II only, the ones with a low Li content between 0 and 0.25 consisted of Phase II and III, with the latter being the dominant phase in the mixture. The samples with an intermediate Li content between 0.25 and 0.71 were mixtures of all three cubic phases. TXRD studies successfully revealed structural evolution of LixMNO as functions of both temperature and Li content. At high temperature, single-phase LixMNO solid solutions formed in Li-rich samples as the miscibility gap between Phase I and II was reduced. The intermediates remained phase pure after cooling to room temperature and their structural similarity to the original spinel Phase I was further confirmed by the vibrational and XAS studies. A new spinel-type impurity phase was detected in samples with x < 0.71, which was attributed to the thermal decomposition products of Phase III. It was speculated that the process releases oxygen from the spinel lattice and triggers nickel reduction to a lower valence state as a compensation mechanism. The chemical nature of the new spinel-type phase, however, is unclear at this point and it is possible that it represents several structurally similar phases and/or compounds. Further characterization is currently underway and the results will be reported in a future publication.
The study reveals that the solid solution behavior in LMNO system bears a great deal of similarity to that of LiFePO4, such as: 1) they are metastable in nature, 2) they can appear as transient intermediates during electrochemical charge and/or discharge and 3) their formation is promoted by temperature. However, LixMNO solid solutions appear to possess higher stability compared to LixFePO4, despite metastable in nature. They are easily detectable by in situ techniques such as XRD and the thermally-driven single phases can be preserved at room temperature. Considering the higher Li mobility in LMNO, the reason for the enhanced stability is unclear and it requires further investigation. Our findings provide pathways for detailed evaluation on the properties of solid solution intermediates, which may further lead to the understanding of their role in the performance of intercalation electrode materials. This study also sets an example for the synthesis of difficult metastable phases that may be used as new electrode materials.
Methods
Unless otherwise specified, all chemicals were obtained from Aldrich with a purity of 97% or higher. Well-formed octahedral-shaped LiMn1.5Ni0.5O4 crystals were prepared according to the procedure described in our previous report17. Various levels of chemical delithiation were achieved by reacting the pristine powder with 0.1 M nitronium tetrafluoroborate in acetonitrile solution in an Argon filled glove box (O2 < 1 ppm and H2O < 1 ppm) at room temperature. The resulting reaction mixtures were filtered, thoroughly washed with acetonitrile and then dried overnight in a vacuum oven. Chemical composition of the samples was determined by an inductively coupled plasma optical emission spectrometer (ICP-OES, Perkin-Elmer Optima 5400). X-ray diffraction patterns were collected using a Panalytical X'Pert Pro diffractometer with monochromatized Cu Kα radiation. The scans were collected between 32 and 80° (2θ) at a rate of 0.0001°/s and a step size of 0.022°. Lattice parameters and phase ratios were determined by full-pattern Rietveld refinements using Riqas software (Materials Data, Inc.). Temperature-controlled XRD studies were carried out in the same diffractometer equipped with an Anton Parr HTK 1200 hot stage. The samples were heated in air at a rate of 5°C/min and the XRD patterns were recorded at a temperature step size of 25°C, with each temperature holding for 5 min before data collection. The same procedure was used for the cooling process. Lattice parameters and phase ratios were determined by full-pattern Rietveld refinements using a Pearson VII function in the RIQAS software (Materials Data, Inc.). The structural model used is  space group with the following Wyckoff position: Li (8a), Ni/Mn (16d) and O (32e). The refined scale factor was used to obtain the phase fractions. The goodness of the fitting is shown in Table 1 in the supporting information. FTIR measurements were performed on KBr pellets using a Nicolet 6700 spectrometer in transmission mode with a spectral resolution of 4 cm−1. Raman spectra of the powder samples were recorded on a “Labram” Raman confocal microscope system (ISA Groupe Horiba) in the confocal backscattering configuration with a 488 nm Argon ion laser (Coherent Inc. Innova 70), a plan olympus 10 × magnification, a 0.25 numerical aperture objective lense and a 22 focal length. Hard XAS data (Mn and Ni K-edges) were collected in transmission mode using a (220) monochromator at SSRL beamline 4-1. Fine powders of as-prepared and heat-treated LixMNO samples were sandwiched between a kapton tap for data collection. Higher harmonics in the X-ray beam were reduced by detuning the Si (220) monochromator by 35% at the Ni edge and 50% at the Mn edge. Energy calibration was accomplished by using the first inflection points in the spectra of Ni and Mn metal foil references, which were 8333 and 6539 eV, respectively. XANES data were analyzed by Sam's Interface for XAS Package or SIXPACK software, with the Photoelectron Energy Origin E0 determined by the first inflection point of the absorption edge jump.
space group with the following Wyckoff position: Li (8a), Ni/Mn (16d) and O (32e). The refined scale factor was used to obtain the phase fractions. The goodness of the fitting is shown in Table 1 in the supporting information. FTIR measurements were performed on KBr pellets using a Nicolet 6700 spectrometer in transmission mode with a spectral resolution of 4 cm−1. Raman spectra of the powder samples were recorded on a “Labram” Raman confocal microscope system (ISA Groupe Horiba) in the confocal backscattering configuration with a 488 nm Argon ion laser (Coherent Inc. Innova 70), a plan olympus 10 × magnification, a 0.25 numerical aperture objective lense and a 22 focal length. Hard XAS data (Mn and Ni K-edges) were collected in transmission mode using a (220) monochromator at SSRL beamline 4-1. Fine powders of as-prepared and heat-treated LixMNO samples were sandwiched between a kapton tap for data collection. Higher harmonics in the X-ray beam were reduced by detuning the Si (220) monochromator by 35% at the Ni edge and 50% at the Mn edge. Energy calibration was accomplished by using the first inflection points in the spectra of Ni and Mn metal foil references, which were 8333 and 6539 eV, respectively. XANES data were analyzed by Sam's Interface for XAS Package or SIXPACK software, with the Photoelectron Energy Origin E0 determined by the first inflection point of the absorption edge jump.
References
Padhi, A. K., Nanjundaswamy, K. & Goodenough, J. B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997).
Thackeray, M., David, W., Bruce, P. & Goodenough, J. B. Lithium insertion into manganese spinels. Mater. Res. Bull. 18, 461–472 (1983).
Ohzuku, T., Ueda, A. & Yamamoto, N. Zero-Strain Insertion Material of Li [Li1/3Ti5/3] O4 for Rechargeable Lithium Cells. J. Electrochem. Soc. 142, 1431–1435 (1995).
Huang, H., Yin, S. C., Kerr, T., Taylor, N. & Nazar, L. F. Nanostructured composites: A high capacity, fast rate Li3V2(PO4)3/carbon cathode for rechargeable lithium batteries. Adv. Mater. 14, 1525–1528 (2002).
Recham, N. et al. Formation of a Complete Solid Solution between the Triphylite and Fayalite Olivine Structures. Chem. Mater. 20, 6798–6809 (2008).
Kobayashi, G. et al. Isolation of Solid Solution Phases in Size-Controlled LixFePO4 at Room Temperature. Adv. Funct. Mater. 19, 395–403 (2009).
Gibot, P. et al. Room-temperature single-phase Li insertion/extraction in nanoscale LixFePO4 . Nat. Mater. 7, 741–747 (2008).
Liu, H. et al. Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes. Science 344 10.1126/science.1252817 (2014).
Delacourt, C., Poizot, P., Tarascon, J. M. & Masquelier, C. The existence of a temperature-driven solid solution in LixFePO4 for 0 ≤ x ≤ 1. Nat. Mater. 4, 254–260 (2005).
Chen, G., Song, X. & Richardson, T. J. Metastable Solid-Solution Phases in the LiFePO4/FePO4 System. J. Electrochem. Soc. 154, A627–A632 (2007).
Kim, J. H., Myung, S. T., Yoon, C. S., Kang, S. G. & Sun, Y. K. Comparative Study of LiNi0.5Mn1.5O4-δ and LiNi0.5Mn1.5O4 Cathodes Having Two Crystallographic Structures: and P4332. Chem. Mater. 16, 906–914 (2004).
Patoux, S. et al. High voltage spinel oxides for Li-ion batteries: From the material research to the application. J. Power Sources 189, 344–352 (2009).
Amine, K., Tukamoto, H., Yasuda, H. & Fujita, Y. A New Three-Volt Spinel Li1 + xMn1. 5Ni0.5O4 for Secondary Lithium Batteries. J. Electrochem. Soc. 143, 1607–1613 (1996).
Zhong, Q., Bonakdarpour, A., Zhang, M., Gao, Y. & Dahn, J. Synthesis and Electrochemistry of LiNixMn2−xO4 . J. Electrochem. Soc. 144, 205–213 (1997).
Ariyoshi, K., Iwakoshi, Y., Nakayama, N. & Ohzuku, T. Topotactic Two-Phase Reactions of Li[Ni1/2Mn3/2]O4 ( P4332 ) in Nonaqueous Lithium Cells. J. Electrochem. Soc. 151, A296–A303 (2004).
Mukerjee, S. et al. In situ synchrotron X-ray studies on copper–nickel 5 V Mn oxide spinel cathodes for Li-ion batteries. Electrochim. Acta 49, 3373–3382 (2004).
Hai, B., Shukla, A. K., Duncan, H. & Chen, G. The effect of particle surface facets on the kinetic properties of LiMn1.5Ni0.5O4 cathode materials. J. Mater. Chem. A. 1, 759–769 (2013).
Cabana, J. et al. Comparison of the Performance of LiNi1/2Mn3/2O4 with Different Microstructures. J. Electrochem. Soc. 158, A997–A1004 (2011).
Ma, X., Kang, B. & Ceder, G. High rate micron-sized ordered LiNi0.5Mn1.5O4 . J. Electrochem. Soc. 157, A925–A931 (2010).
Shaju, K. M. & Bruce, P. G. Nano-LiNi0.5Mn1.5O4 spinel: a high power electrode for Li-ion batteries. Dalton Trans. 40, 5471–5475 (2008).
Xiao, J. et al. Interplay between two-phase and solid solution reactions in high voltage spinel cathode material for lithium ion batteries. J. Power Sources 242, 736–741 (2013).
Arunkumar, T. A. & Manthiram, A. Influence of Lattice Parameter Differences on the Electrochemical Performance of the 5 V Spinel LiMn1.5−yNi0.5−zMy + zO4 (M = Li, Mg, Fe, Co and Zn). Electrochem. Solid-State Lett. 8, A403–A405 (2005).
Cabana, J. et al. Composition-Structure Relationships in the Li-Ion Battery Electrode Material LiNi0.5Mn1.5O4 . Chem. Mater. 24, 2952–2964 (2012).
Amdouni, N., Zaghib, K., Gendron, F., Mauger, A. & Julien, C. M. Structure and insertion properties of disordered and ordered LiNi0.5Mn1.5O4 spinels prepared by wet chemistry. Ionics 12, 117–126 (2006).
Lee, E. & Persson, K. A. Solid-Solution Li Intercalation as a Function of Cation Order/Disorder in the High-Voltage LixNi0.5Mn1.5O4 Spinel. Chem. Mater. 25, 2885–2889 (2013).
Wang, L., Li, H., Huang, X. & Baudrin, E. A comparative study of Fd-3m and P4332 “LiNi0.5Mn1.5O4”. Solid State Ionics 193, 32–38 (2011).
Rhodes, K., Meisner, R., Kim, Y., Dudney, N. & Daniel, C. Evolution of Phase Transformation Behavior in Li(Mn1.5Ni0.5)O4 Cathodes Studied By In Situ XRD. J. Electrochem. Soc. 158, A890–A897 (2011).
Kunduraci, M. & Amatucci, G. G. Synthesis and Characterization of Nanostructured 4.7 V LixMn1.5Ni0.5O4 Spinels for High-Power Lithium-Ion Batteries. J. Electrochem. Soc. 153, A1345–A1352 (2006).
Zheng, J. et al. Enhanced Li+ ion transport in LiNi 0.5Mn1.5O4 through control of site disorder. PCCP 14, 13515–13521 (2012).
Kim, J.-H., Yoon, C. S., Myung, S.-T., Prakash, J. & Sun, Y.-K. Phase Transitions in Li1 −δNi0.5Mn1.5O4 during Cycling at 5 V. Electrochem. Solid-State Lett. 7, A216–A220 (2004).
Hu, E. et al. Oxygen-Release-Related Thermal Stability and Decomposition Pathways of LixNi0.5Mn1.5O4 Cathode Materials. Chem. Mater. 26, 1108–1118 (2013).
Julien, C. M. & Massot, M. Lattice vibrations of materials for lithium rechargeable batteries I. Lithium manganese oxide spinel. Mater. Sci. Eng. B. 97, 217–230 (2003).
Desilvestro, J., Corrigan, D. A. & Weaver, M. J. Spectroelectrochemistry of thin nickel hydroxide films on gold using surface-enhanced Raman spectroscopy. J. Phys. Chem. 90, 6408–6411 (1986).
Wei, Y., Kim, K.-B. & Chen, G. Evolution of the local structure and electrochemical properties of spinel LiNixMn2−xO4 (0 ≤ x ≤ 0.5). Electrochim. Acta. 51, 3365–3373 (2006).
Baddour-Hadjean, R. & Pereira-Ramos, J. P. Raman Microspectrometry Applied To The Study Of Electrode Materials For Lithium Batteries. AIP Conf. Proc. 1267, 1137–1138 (2010).
Jin, S. J. et al. Effect of composition change of metals in transition metal sites on electrochemical behavior of layered Li[Co1−2x(Li1/3Mn2/3)x(Ni1/2Mn1/2)x]O2 solid solutions. Solid State Ionics 177, 105–112 (2006).
Talyosef, Y. et al. The study of LiNi0.5Mn1.5O4 5-V cathodes for Li-ion batteries. J. Power Sources 146, 664–669 (2005).
Terada, Y. et al. In Situ XAFS Analysis of Li(Mn,M)2O4 (M = Cr, Co, Ni) 5V Cathode Materials for Lithium-Ion Secondary Batteries. J. Solid State Chem. 156, 286–291 (2001).
Nam, K.-W. et al. Combining In Situ Synchrotron X-Ray Diffraction and Absorption Techniques with Transmission Electron Microscopy to Study the Origin of Thermal Instability in Overcharged Cathode Materials for Lithium-Ion Batteries. Adv. Funct. Mater. 23, 1047–1063 (2013).
Arai, H. et al. Phase transition kinetics of LiNi0.5Mn1.5O4 electrodes studied by in situ X-ray absorption near-edge structure and X-ray diffraction analysis. J. Mater. Chem. A. 1, 10442–10449 (2013).
Kim, M. G. & Yo, C. H. X-ray Absorption Spectroscopic Study of Chemically and Electrochemically Li Ion Extracted LiyCo0.85Al0.15O2 Compounds. J. Phys. Chem. B. 103, 6457–6465 (1999).
Ignatov, A. Y., Ali, N. & Khalid, S. Mn K-edge XANES study of the La1-xCaxMnO3 colossal magnetoresistive manganites. Phys. Rev. B 64, 014413 (2001).
Kim, M. G., Shin, H. J., Kim, J.-H., Park, S.-H. & Sun, Y.-K. XAS Investigation of Inhomogeneous Metal-Oxygen Bond Covalency in Bulk and Surface for Charge Compensation in Li-Ion Battery Cathode Li[Ni1/3Co1/3Mn1/3]O2 Material. J. Electrochem. Soc. 152, A1320–A1328 (2005).
Acknowledgements
The authors acknowledge the support of Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. We thank Drs. Marca Doeff, Feng Lin, Ryan Davis, Doug Van Campen and Chad Miller for assisting with the synchrotron experiments and Drs. Thomas Richardson and Hugues Duncan for helpful discussion. This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of FreedomCAR and Vehicle Technologies of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
S.K. prepared the samples, performed the experiemental studies and prepared the figures under the supervision of G.C. S.K. and G.C. wrote the main manuscript text. A.J. and R.K. performed Raman characterization of the samples and contributed to writing the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
A study of room-temperature LixMn1.5Ni0.5O4 solid solutions
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Saravanan, K., Jarry, A., Kostecki, R. et al. A study of room-temperature LixMn1.5Ni0.5O4 solid solutions. Sci Rep 5, 8027 (2015). https://doi.org/10.1038/srep08027
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08027
This article is cited by
-
Propagation topography of redox phase transformations in heterogeneous layered oxide cathode materials
Nature Communications (2018)
-
Phase transformation mechanism in lithium manganese nickel oxide revealed by single-crystal hard X-ray microscopy
Nature Communications (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
