
Abstract
Here, we report computational studies of the SH3 protein domain interacting with various single-walled carbon nanotubes (SWCNT) either bare or functionalized by mimicking the proline-rich motif (PRM) ligand (PPPVPPRR) and compare it to the SH3-PRM complex binding. With prolines or a single arginine attached, the SWCNT gained slightly on specificity when compared with the bare control, whereas with multi-arginine systems the specificity dropped dramatically to our surprise. Although the electrostatic interaction provided by arginines is crucial in the recognition between PRM and SH3 domain, our results suggest that attaching multiple arginines to the SWCNT has a detrimental effect on the binding affinity. Detailed analysis of the MD trajectories found two main factors that modulate the specificity of the binding: the existence of competing acidic patches at the surface of SH3 that leads to “trapping and clamping” by the arginines and the rigidity of the SWCNT introducing entropic penalties in the proper binding. Further investigation revealed that the same “clamping” phenomenon exits in the PRM-SH3 system, which has not been reported in previous literature. The competing effects between nanoparticle and its functionalization components revealed by our model system should be of value to current and future nanomedicine designs.
Similar content being viewed by others
Introduction
Conventional drugs often suffer from low or limited sustained efficacy due to immune degradation and lack of specificity1. In contrast, nanomedicines, such as SWCNT functionalized with various biomolecules, may survive immune clearance and target malignant cells, making them very promising nanovectors2 and nanovaccines3. Examples include SWCNT-PTX conjugates for tumor suppression4 and carborane-appended SWCNTs for boron neutron capture therapy5.
The nanotoxicity of a bare prototypical carrier is, however, a big concern6,7,8,9,10. It has been reported that bare SWCNT may induce very strong biotoxicity at different scales. For example, at the molecular level it can plug into the hydrophobic core of WW domains and disrupt their active sites6. Its toxicity has also been identified at the cellular and organ level through various experiments11,12,13. Other nanomaterials such as graphene share similar toxicity–recent experiments showed that pristine graphene and graphene oxide nanosheets can induce the degradation of the inner and outer cell membranes of Escherichia coli, thus reducing their viability14,15,16,17. As a result, nanotoxicity alleviation has emerged as an important research direction for devising safer ways to wield these man-made particles.
The Src homology 3 (SH3) protein domain is usually found at the downstream of signal transduction pathways and plays an important role mediating protein-protein interactions18. This protein domain is known to bind with high specificity to sequences rich in proline and other hydrophobic amino acids. One of such ligands, the proline-rich motif (PRM, peptide sequence PPPVPPRR)19 binds to the SH3 domain by following a two-step process20. First, the two positively charged arginines at the tail of the PRM interact with the negatively charged acidic patches around the binding pocket. This long-range electrostatic attraction connects the tail of the PRM to the acidic patch, thus effectively reducing the overall search space. Once anchored, hydrophobic interactions between the PRM and the binding pocket induce a local interfacial drying through strong hydrophobic packing, thus fixating the PRM into the binding pocket.
Interestingly, recent studies showed that bare SWCNTs could compete with PRM in binding to SH3 domains7, which may in turn interrupt the signal transduction and thus disrupt the biological function of SH3 domain, ultimately leading to toxic effects.
Meanwhile, the technology for carbon nanotube functionalization has made significant progress in the past decade. Strano et al.21 found that diazonium reagents could functionalize single-walled carbon nanotubes suspended in aqueous solution, achieving high selectivity and controllability. Banerjee and his colleagues22 investigated the strategy of molecular metal complexation with SWCNTs to control site-selective chemistry in the functionalization. These recent promising technologies for custom-functionalizing SWCNTs can be leveraged to investigate how different functionalizations affect the nanotoxicity of the nano structures, with an ultimate goal of reducing or completely eliminating the adverse effects of cell exposure to nanomaterials. On the other hand, molecular dynamics (MD) simulations are a remarkable tool for the detailed study of objects in nanoscales. MD has been widely used in areas such as protein folding23,24,25,26,27,28,29,30, ligand-receptor binding31,32,33,34, nanotoxicity8,14,35, nanomedicine14,35,36 and nanomachine development37,38,39.
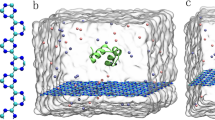
In this study, we have functionalized short (3,3) SWCNTs by bio-mimicking the PRM in four different models and compare their binding with PRM and bare SWCNT. Since the prolines on PRM provide hydrophobic interactions required for specific binding, we attached 3 key proline residues40 to the SWCNT according to their relative position on the PRM (system referred as P3). Similarly, given that the arginines play a crucial role in the initial SH3 domain recognition, we attached arginines to the tail of the SWCNT in 3 different scaffolds: one single arginine (system A1), two linked arginines (system A2l) and two separate arginines (system A2s) [Figure 1]. The results of each functionalization are then compared to a control simulation consisting of a bare SWCNT (bare) and to another control simulation containing only the PRM (PRM).

Six different “ligands” were simulated in presence of a SH3 protein domain.
First two are the control systems–(a) the native ligand PRM and (b) bare SWCNT. The rest are functionalized SWCNT systems, (c) P3–SWCNT with three prolines (d) A1–SWCNT with one arginine (e) A2l–SWCNT with two linked arginines and (f) A2s–SWCNT with two separate arginines.
We found that the functionalization strategies employed here generally failed to meaningfully improve the specificity of the SWCNT-SH3 interaction, regardless of our great efforts in bio-mimicking PRM faithfully. In some extreme cases, such as some arginine-based functionalization, it even reduced the specificity significantly. These surprising results suggest that the prevalence of intricate subtleties underlying nanoparticle functionalization remains a major concern in today's nanomedicine design. Moreover, the same mechanism leads to the failure of fCNT was also observed in the PRM-SH3 system and reported here for the first time. However, in this case, the biological peptide has the flexibility to offset most of the negative effect. Despite the unfavorable and unexpected outcomes, by elucidating the mechanism behind this specificity loss, our current findings may help to identify potential new strategy to mitigate the risks of nanotoxicity in nanomedicine design.
Results and Discussion
We carried out 10 × 100 ns simulations for each of the four fCNT models mentioned above, along with the two control systems (CNT and PRM), in the presence of the SH3 domain (total six systems). In general, a sufficient sampling is necessary for any meaningful conclusions. However, to guarantee a perfect ergodic sampling is very hard or impossible for complex biological systems49,50. In this particular case, we performed 10 independent trajectories for each case to collect data, which is better or comparable to many similar studies in literature. The Cartesian distance between the geometrical center of the binding pocket (defined as β-carbons of relevant residues of F8, W36, P50 and Y53)41 and the center of the fCNT (DPC), along with another distance between the center of the binding pocket and the longest end (tip) of the fCNT (DPE), were monitored to provide a comprehensive description of the binding process.
Free energy landscapes (FEL) of all different systems using the reaction coordinates mentioned above display successful binding, marked by a large barrier separating “bound” from “unbound” on each case [Figure 2]. The bound state is located in the lower-left corner and labeled with a “B”, while a more extended and rough “not bound” basin spans the middle of the landscape.

Free energy landscapes of (a) Bare SWCNT (b) SWCNT with 3 prolines (c) SWCNT with one arginine (d) SWCNT with two linked arginines (e) SWCNT with two separated arginines.
The reaction coordinate consists of the distance between the SWCNT center and binding pocket as the abscissa and the distance between the SWCNT tip and binding pocket as the ordinate. The lower-left areas marked with “B” represents the bound state. For the first four systems the bound state is rather competitive, but for (e) A2s it has shrunk and shallowed significantly, which is consistent with the low binding success rate.
Comparison of all six systems demonstrate that the bound basins in SWCNT, P3, A1 and A2l all compete against the non-bound basins, though they are all comparable with each other with no favor for the functionalized systems. On the other hand, the bounded basin in A2s system shows a radical recession in both size and depth, making it a shallow local minimum. These observations indicate that the functionalization has very limited positive contribution to the recognition between SWCNT and SH3 binding pocket and some may even cause significant loss of specificity.
The bound and unbound basins are well separated in all cases which suggests Dpc = 1.3 nm as an adequate cut-off distance to distinguish the bound and unbound states in the time scales sampled. Hence, we took the average  of the last 5 ns for each trajectory as an indicator for its final binding status, with
of the last 5 ns for each trajectory as an indicator for its final binding status, with  nm considered bound. We then define the binding success rate of each system as rb = Nsuccess/Nall, which serves as a direct proxy to measure the binding specificity for each system.
nm considered bound. We then define the binding success rate of each system as rb = Nsuccess/Nall, which serves as a direct proxy to measure the binding specificity for each system.
While the bare SWCNT is not guided by any electrostatic interaction, surprisingly, the super-hydrophobic SWCNT still provides high specificity to the binding pocket. The bound basin is the global minimum of the free energy landscape, with rb = 3/10. Interestingly, due to the high hydrophobicity of the binding pocket, when the first contact between the SH3 domain and the bare SWCNT was not in the binding site, the nanotube was able to roll to the proper position [Figure S1]. This “rolling” can also be seen in its FEL [Figure 2(a)], whose inverted L shape suggests SWCNT usually first moves in the Dpc direction (find the pocket), then in the Dpe direction (adjust position) to reach the bound state.
In turn, the P3 system displayed a slight boost on the binding success rate, managing to reach rb = 4/10. The extremely hydrophobic SWCNT competed against the prolines for binding to the protein domain, rendering the fCNT only slightly more specific than the control system. Visual inspection of the trajectories showed that steric effects were the main contributor to the disruption of better binding. In our simulations, when the prolines made contact with the binding pocket, the nearby SWCNT carbon rings would have hard time making their own contacts with the pocket due to the rigidity of CNT. This can also be seen in the FEL in terms of the separation of both the bound and unbound basins, in smaller mutually exclusive local minima. This is further observed in the free energy landscape of P3 with the distance between prolines and binding pocket as second reaction coordinate [Figure S2], where the bound basin is somewhat divided diagonally, depending on whether the prolines or SWCNT occupy the pocket.
The functionalized SWCNT with a single arginine (A1) displayed the same binding success rate as P3. Even though steric effects were not observed in the simulated trajectories, the existence of other acidic patches with comparable size to the one near the binding pocket (the right one for PRM, see Figure 3) provided the single arginine some competing sites for the long-range recognition. These surface charges effectively roughened the free energy landscape of binding by introducing competing local minima. While modest, the inclusion of a single arginine did improve the SH3 recognition when compared to the control system.

Screenshots of one representative trajectory of the A2s system (SWCNT with two linked arginines) which shows successfully binding to the SH3 domain.
The acidic residues and functionalized arginines are in Licorice representation, while key binding pocket residues are shown as VDW spheres. (1) At first fCNT was 30  away from the SH3 domain. (2) fCNT quickly found an acidic patch and clamps to it in 6 ns. (3) At 44 ns the clamping broke and (4) fCNT were docking onto the binding pocket sideway. (5) Two arginines made contact with two acidic patches respectively and the entire complex stayed stable to the end of the simulation.
away from the SH3 domain. (2) fCNT quickly found an acidic patch and clamps to it in 6 ns. (3) At 44 ns the clamping broke and (4) fCNT were docking onto the binding pocket sideway. (5) Two arginines made contact with two acidic patches respectively and the entire complex stayed stable to the end of the simulation.
Inclusion of a second arginine in the A2l system, which bio-mimics exactly the original arginines of the PRM, was tried next. To our surprise, with this exact-mimicking strategy providing doubled electrostatic interaction the binding success rate dropped to r = 3/10. Visual inspection of the trajectories revealed that the second arginine allowed for a newly emerging “clamping mechanism”. While in the single arginine case the binding between arginine and the competing charged patches was short-lived, the doubly functionalized A2l could clamp around the patch [Figure 3], resulting in very effective trapping with much longer residing time. This mechanism explains the reduced binding specificity observed. In a representative trajectory where A2l successfully located the binding pocket [Figure 3], we observed a rapid establishment of clamping at around 6 ns. The fCNT was trapped there for about 40 ns, then the clamping contact broke and fCNT moved closer to the binding pocket. In the following 50 ns, it was constantly adjusting its position, until 94 ns into the simulation, when its two arginines managed to make direct contact with the two acidic patches respectively.
Guided by the observations described above, we decided to utilize and maximize the clamping effect by attaching two separate arginines to the SWCNT (A2s). This time the clamping was greatly favored by the flexibility of the two distant arginine arms, with 5 out of 10 trajectories adopting the clamping scenario and keeping it to the end of the simulation, decreasing the binding success rate to r = 1/10. Figure S3 shows the free energy landscape with the distance between the nitrogen atoms of the two arginine arms and the distance between the pocket and fCNT as reaction coordinates. When projected to this new reaction coordinate with the distance between two Arg arms, it is easy to see the bound basin displays a 0.5 nm larger separation in the two Arg arms than the non-bound basin, meaning in the unbound state the arginine arms are much closer to each other [Figure S3]. In a typical trajectory, the fCNT quickly finds some acidic patches and stays there [Figure S4], which corresponds to the two arginine arms clamping around respective patches simultaneously [Figure S5]. Meanwhile, the SWCNT, dragged by the two arginines, also gets trapped far away from the binding pocket. All these processes finished in less than 10 ns. The clamped complex was also very stable, remaining unchanged for the next 90 ns.
To further validate the clamping mechanism and given that A2l was functionalized to mimic exactly the PRM arginines, simulations of the PRM peptide in presence of the SH3 domain were performed in order to assess whether this clamping mechanism is also present in the biological system. Interestingly, the simulations showed that PRM also displayed clamping in 3 out of 10 simulations [Figure 4]. However, the flexible backbone of the peptide allowed the PRM to curl itself in order for the prolines to find the pocket. This flexibility contrasts drastically with the case of fCNT, where even clamping in the vicinity of the pocket prevents successful binding.

The tail of PRM (blue) is clamping around an acidic patch, but the main body (brown) manages to curl itself and find the binding pocket.
Based on all the results we had obtained, we infer that there are inherent subtleties underlying nanoparticle functionalization. This is instructive for the future design of more complicated and realistic systems.
First, the functionalization of nanoparticles must adopt a holistic view. Today's nanomedicines with specific targeting capability are often functionalized with multiple components to be multi-functional, which would enable them to work properly all the way from administration to drug action2. These multiple components should be designed to work cooperatively with each other, which could be a big challenge as illustrated by the P3 system. Our systems indicated that the flexibility of the substrate material, modification pattern and the local environment of target protein surface were all important factors.
Second, exact mimicking of the biological system will not necessarily yield optimal design. Bio-mimicking is very prevalent in today's design of high efficacy anti-cancer drugs, like tagging the nanocarrier with specific antibodies3 or other small naturally arising ligands42. From our model system, we observe that the original mechanism may not work well with the new substrate nanoparticle. Those direct bio-mimicking products potentially need to be improved further by carefully investigating and utilizing the molecular level interactions in their specific environment as a whole.
Our approach for the A2s design is reminiscent of the “negative design” in the field of protein design, where the unfavorable interactions are removed to make the protein folding smoothely43, while here we were enhancing the competing interaction to alleviate the nanotoxicity. This is different from the previous nanotoxicity alleviation methods, which often adopt the strategy of armoring SWCNTs with other residues or functional groups by covalent bonding (most frequently small hydrophilic groups like –OH and –NH3), or proteins by non-covalent interaction8.
Finally, it should be noted that pristine SWCNTs are highly insoluble in water and thus not accessible to biomolecules in solution. With sufficient functionalization to be water dispersible, it is then important to make sure that the modified SWCNT still has large enough exposed hydrophobic surface to bind to the pocket of SH3 domain. Fortunately, this seems not a problem at all. In a previous study on SWCNT interacting with human serum proteins8, we found that functionalized SWCNTs (so water dispersible) still have large exposed hydrophobic surface areas that can absorb large amount of serum proteins. In this particular case, we coat the (small-sized) SWCNT with multiple charged residues, which should help the functionalized SWCNT to be water dispersible, while still maintaining a large exposed hydrophobic surface area.
Conclusion
We have carried out atomistic molecular dynamics simulations for bare SWCNT, PRM and four different fCNTs interacting with a SH3 domain in explicit water. The SWCNTs were functionalized with three prolines (P3), a single arginine (A1), two consecutive arginines (A2l) and two separate arginines (A2s) respectively and the resulting binding properties were compared. For P3 and A1, the binding success rate only improved slightly from 3/10 to 4/10. Our analysis showed that steric effects hindered the specificity of P3, while the competing interaction from non-relevant acidic patches lowered the possibility of better binding in A1 system. To our surprise, the specificity dropped back to 3/10 and 1/10 for the multi-arginine systems, regardless of their increased electrostatic attraction and high resemblance to the PRM. The deficient binding of the A2l and A2s systems were due to the emerging “clamping mechanism” along with the rigidity of SWCNT. This raised the question of whether or not the clamping exists in the original biological system (PRM-SH3), which was then confirmed in our simulation.
These subtleties raised questions about the linear thinking (interpolation) and faithful mimicking of biological system in nanoparticle functionalization. A systematic view must be adopted, especially in the design of multi-functional nano-systems, where the cooperation between different components is crucial.
Methods
We used a 19.54  long, (3,3) SWCNT in all our simulations, generated with the VMD 1.9.1 package44. The small (3,3) nanotube was used mainly to mimic the size of the natural proline-rich motif peptide in order to better fit into the hydrophobic groove of SH3 domain. A slightly larger SWCNT, such as (4,4), (5,5) or (6,6), can in theory also work, maybe to a less extent. In addition to size, the surface curvature can also play a meaningful role as indicated in recent studies35,51. The carbon atoms were modeled as uncharged Lennard-Jones particles with a depth of potential well of
long, (3,3) SWCNT in all our simulations, generated with the VMD 1.9.1 package44. The small (3,3) nanotube was used mainly to mimic the size of the natural proline-rich motif peptide in order to better fit into the hydrophobic groove of SH3 domain. A slightly larger SWCNT, such as (4,4), (5,5) or (6,6), can in theory also work, maybe to a less extent. In addition to size, the surface curvature can also play a meaningful role as indicated in recent studies35,51. The carbon atoms were modeled as uncharged Lennard-Jones particles with a depth of potential well of  and a cross section of σcc = 3.40
and a cross section of σcc = 3.40  45. The structure of SH3 domain and PRM ligand were downloaded from Protein Data Bank (PDB code: 1CKB40). The fCNTs were positioned 30
45. The structure of SH3 domain and PRM ligand were downloaded from Protein Data Bank (PDB code: 1CKB40). The fCNTs were positioned 30  away from the SH3 domain in all the systems and the initial relative position was chosen at random for each run.
away from the SH3 domain in all the systems and the initial relative position was chosen at random for each run.
To carry out the molecular dynamics simulation, we used the Gromacs package 4.5.546. All the systems were modeled with the OPLSAA force field47and solvated in cubic periodic boxes filled with around 12,000 TIP3P water molecules48, with the minimum distance between solutes and the box boundary set as 10  . We then added Na+ to balance the charge and carried energy minimization. After that, we equilibrated all the systems at a 1 bar pressure and 300 K temperature for 400 ps using Berendsen coupling. The production run were performed at 300 K with a time step of 2 fs, LINCS algorithm and particle-mesh Ewald method (PME) were adopted and the van der Waals interactions were treated with a smooth cutoff distance set to 12
. We then added Na+ to balance the charge and carried energy minimization. After that, we equilibrated all the systems at a 1 bar pressure and 300 K temperature for 400 ps using Berendsen coupling. The production run were performed at 300 K with a time step of 2 fs, LINCS algorithm and particle-mesh Ewald method (PME) were adopted and the van der Waals interactions were treated with a smooth cutoff distance set to 12  . For each system we obtained 10 trajectories with 100 ns in length.
. For each system we obtained 10 trajectories with 100 ns in length.
References
Nel, A. E. et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 8, 543–557 (2009).
Ferrari, M. Cancer nanotechnology: opportunities and challenges. Nat. Rev. Cancer. 5, 161–171 (2005).
Bianco, A., Kostarelos, K. & Prato, M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 9, 674–679 (2005).
Liu, Z. et al. Drug Delivery with Carbon Nanotubes for In vivo Cancer Treatment. Cancer. Res. 68, 6652–6660 (2008).
Yinghuai, Z., Peng, A. T. & Carpenter, K. Substituted carborane-appended water-soluble single-wall carbon nanotubes: new approach to boron neutron capture therapy drug delivery. J. Am. Chem. Soc. 127, 9875–9880 (2005).
Zuo, G., Huang, Q., Wei, G., Zhou, R. & Fang, H. Plugging into proteins: poisoning protein function by a hydrophobic nanoparticle. Acs. Nano. 4, 7508–7514 (2010).
Zuo, G., Gu, W., Fang, H. & Zhou, R. Carbon nanotube wins the competitive binding over proline-rich motif ligand on sh3 domain. J. Phys. Chem. C. 115, 12322–12328 (2011).
Ge, C. et al. Binding of blood proteins to carbon nanotubes reduces cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 108, 16968–16973 (2011).
Braydich-Stolle, L. K. et al. Crystal structure mediates mode of cell death in TiO2 nanotoxicity. J. Nanopart. Res. 11, 1361–1374 (2009).
Fischer, H. C. & Chan, W. C. Nanotoxicity: the growing need for in vivo study. Curr. Opin. Biotech. 18, 565–571 (2007).
Smith, C. J., Shaw, B. J. & Handy, R. D. Toxicity of single walled carbon nanotubes to rainbow trout, (Oncorhynchus mykiss): Respiratory toxicity, organ pathologies and other physiological effects. Aquat. toxicol. 82, 94–109 (2007).
Magrez, A., Kasas, S., Salicio, V., Pasquier, N. & Seo, J. W. Cellular toxicity of carbon-based nanomaterials. Nano. Lett. 6, 1121–1125 (2006).
Davoren, M., Herzog, E., Casey, A. & Cottineau, B. In vitro toxicity evaluation of single walled carbon nanotubes on human A549 lung cells. Toxicol. Vitro. 21, 438–448 (2007).
Tu, Y. et al. Destructive extraction of phospholipids from Escherichia coli membranes by graphene nanosheets. Nat. Nanotechnol. 8, 594–601 (2013).
Hu, W. et al. Graphene-Based Antibacterial Paper. Acs. Nano. 4, 4317–4323 (2010).
Liu, S. et al. Antibacterial Activity of Graphite, Graphite Oxide, Graphene Oxide and Reduced Graphene Oxide: Membrane and Oxidative Stress. Acs. Nano. 5, 6971–6980 (2011).
Akhavan, O. & Ghaderi, E. Toxicity of graphene and graphene oxide nanowalls against bacteria. Acs. Nano. 4, 5731–5736 (2010).
Pawson, T. & Nash, P. Assembly of Cell Regulatory Systems Through Protein Interaction Domains. Science 300, 445–452 (2003).
Smithgall, T. E. SH2 and SH3 domains: potential targets for anti-cancer drug design. J. Pharmacol. Toxicol. Methods. 34, 125–132 (1995).
Ahmad, M., Gu, W. & Helms, V. Mechanism of Fast Peptide Recognition by SH3 Domains. Angew. Chem. Int. Ed. 47, 7626–7630 (2008).
Strano, M. S. et al. Electronic structure control of single-walled carbon nanotube functionalization. Science 301, 1519–1522 (2003).
Banerjee, S., Kahn, M. & Wong, S. S. Rational chemical strategies for carbon nanotube functionalization. Chem. Eur. J. 9, 1899–1908 (2003).
Sugita, Y. & Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151 (1999).
Levitt, M. Protein folding by restrained energy minimization and molecular dynamics. J. Mol. Biol. 170, 723–764 (1983).
Snow, C. D., Nguyen, H., Pande, V. S. & Gruebele, M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420, 102–106 (2002).
Zhou, R. Trp-cage: Folding free energy landscape in explicit water. Proc. Natl. Acad. Sci. U. S. A. 100, 13280–13285 (2003).
Zhou, R., Huang, X., Margulis, C. J. & Berne, B. J. Hydrophobic Collapse in Multidomain Protein Folding. Science 305, 1605–1609 (2004).
Liu, P., Huang, X., Zhou, R. & Berne, B. J. Hydrophobic aided replica exchange: an efficient algorithm for protein folding in explicit solvent. J. Phys. Chem. C. 110, 19018–19022 (2006).
Zhou, R., Eleftheriou, M., Royyuru, A. K. & Berne, B. J. Destruction of long-range interactions by a single mutation in lysozyme. Proc. Natl. Acad. Sci. U. S. A. 104, 5824–5829 (2007).
Eleftheriou, M., Germain, R. S., Royyuru, A. K. & Zhou, R. Thermal denaturing of mutant lysozyme with both the OPLSAA and the CHARMM force fields. J. Am. Chem. Soc. 128, 13388–13395 (2006).
Xia, Z. et al. The complex and specific pMHC interactions with diverse HIV-1 TCR clonotypes reveal a structural basis for alterations in CTL function. Sci. Rep. 4 (2014).
Zhou, R., Das, P. & Royyuru, A. K. Single mutation induced H3N2 hemagglutinin antibody neutralization: a free energy perturbation study. J. Phys. Chem. B. 112, 15813–15820 (2008).
Das, P., Li, J., Royyuru, A. K. & Zhou, R. Free energy simulations reveal a double mutant avian H5N1 virus hemagglutinin with altered receptor binding specificity. J. Comput. Chem. 30, 1654–1663 (2009).
Xia, Z., Huynh, T., Kang, S.-G. & Zhou, R. Free-energy simulations reveal that both hydrophobic and polar interactions are important for influenza hemagglutinin antibody binding. Biophys. J. 102, 1453–1461 (2012).
Zuo, G. Zhou, X., Huang, Q., Fang, H. & Zhou, R. Adsorption of villin headpiece onto graphene, carbon nanotube and C60: effect of contacting surface curvatures on binding affinity. J. Phys. Chem. C. 115, 23323–23328 (2011).
Kang, S.-G. et al. Molecular mechanism of pancreatic tumor metastasis inhibition by Gd@C82(OH)22 and its implication for de novo design of nanomedicine. Proc. Natl. Acad. Sci. U. S. A. 109, 15431–15436 (2012).
Li, J. et al. Electrostatic gating of a nanometer water channel. Proc. Natl. Acad. Sci. U. S. A. 104, 3687–3692 (2007).
Lu, H., Zhang, Y., Huynh, T. & Zhou, R. Capability of charge signal conversion and transmission by water chains confined inside Y-shaped carbon nanotubes. J. Chem. Phys. 138, 015104 (2013).
Tu, Y. et al. Water-mediated signal multiplication with Y-shaped carbon nanotubes. Proc. Natl. Acad. Sci. U. S. A. 106, 18120–18124 (2009).
Wu, X., Knudsen, B., Feller, S. M., Zheng, J. & Sali, A. Structural basis for the specific interaction of lysine-containing proline-rich peptides with the N-terminal SH3 domain of c-Crk. Structure 3, 215–226 (1995).
Nguyen, J. T., Turck, C. W., Cohen, F. E., Zuckermann, R. N. & Lim, W. A. Exploiting the Basis of Proline Recognition by SH3 and WW Domains: Design of N-Substituted Inhibitors. Science 282, 2088–2092 (1998).
Henne, W. A., Doorneweerd, D. D. & Hilgenbrink, A. R. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg. Med. Chem. Lett. 16, 5350–5355 (2006).
Koga, N. et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graphics Modell. 14, 33–38 (1996).
Hummer, G., Rasaiah, J. C. & Noworyta, J. P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 414, 188–190 (2001).
Pronk, S., Páll, S., Schulz, R., Larsson, P., Bjelkmar, P., Apostolov, R., Shirts, M. R., Smith, J. C., Kasson, P. M., van der Spoel, D., Hess, B. & Lindahl, E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Duan, Y. et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 (2003).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Straub, J. E. & Thirumalai, D. Theoretical probes of conformational fluctuations in S-peptide and RNase A/3′–UMP enzyme product complex. Proteins. 15, 360–373 (1993).
Zhou, R. & Berne, B. J. Smart walking: A new method for Boltzmann sampling of protein conformations. J. Chem. Phys. 107, 9185–9196 (1997).
Giuseppina, R. & Fabio, G. Surface Topography Effects in Protein Adsorption on Nanostructured Carbon Allotropes. Langmuir. 29, 4883–4893 (2013).
Acknowledgements
We thank Seung-gu Kang, Tien Huynh, Peng Xiu, Xiaowei Tang and Bruce Berne for useful discussions and insightful comments. This work is partially supported by National Natural Science Foundation of China (grant Nos. 21320102003, 11204269). R.Z. acknowledges the financial support from the IBM BlueGene Science Program. A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
R.Z. conceived and designed the research. Y.Z., C.A.J., B.Z. and R.Z. co-wrote the manuscript. Y.Z., B.Z. and Z.Y. carried out the molecular dynamics simulations. Y.Z., C.A.J., B.Z., J.W. and R.Z. analyzed the data and Y.Z. and C.A.J. prepared all the figures. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhang, Y., Jimenez-Cruz, C., Wang, J. et al. Bio-mimicking of Proline-Rich Motif Applied to Carbon Nanotube Reveals Unexpected Subtleties Underlying Nanoparticle Functionalization. Sci Rep 4, 7229 (2014). https://doi.org/10.1038/srep07229
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07229
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
