
Abstract
Transgenic plants producing insecticidal proteins from the bacterium Bacillus thuringiensis (Bt) are useful for pest control, but their efficacy is reduced when pests evolve resistance. Here we examined the mechanism of resistance to Bt toxin Cry1Ac in the laboratory-selected LF5 strain of the cotton bollworm, Helicoverpa armigera. This strain had 110-fold resistance to Cry1Ac protoxin and 39-fold resistance to Cry1Ac activated toxin. Evaluation of five trypsin genes revealed 99% reduced transcription of one trypsin gene (HaTryR) was associated with resistance. Silencing of this gene with RNA interference in susceptible larvae increased their survival on diets containing Cry1Ac. Bioassays of progeny from crosses revealed that resistance to Cry1Ac was genetically linked with HaTryR. We identified mutations in the promoter region of HaTryR in the resistant strain. In transfected insect cell lines, transcription was lower when driven by the resistant promoter compared with the susceptible promoter, implicating cis-mediated down-regulation of HaTryR transcription as a mechanism of resistance. The results suggest that H. armigera can adapt to Bt toxin Cry1Ac by decreased expression of trypsin. Because trypsin activation of protoxin is a critical step in toxicity, transgenic plants with activated toxins rather than protoxins might increase the durability of Bt crops.
Similar content being viewed by others


Introduction
The gram-positive bacterium Bacillus thuringiensis (Bt) produces a variety of crystalline (Cry) proteins that can kill certain insect pests, but have little or no toxicity to most non-target organisms1,2. In 2013, farmers planted transgenic crops producing Bt toxins on 76 million ha worldwide3, but the evolution of resistance in some pest populations has reduced the efficacy of these Bt crops4. Previously reported mechanisms of resistance to Bt toxins include reduced binding of activated Bt toxins to midgut receptors and reduced conversion of Bt protoxins to activated toxins by insect midgut proteases such as trypsin2,5,6,7.
In China, planting of transgenic cotton that produces Bt toxin Cry1Ac has helped to suppress the cotton bollworm, Helicoverpa armigera and reduce insecticide use against this pest8,9. Although Bt cotton has remained useful against H. armigera in China, high levels of resistance to Cry1Ac have been selected in many laboratory strains of H. armigera and significant increases in the frequency of resistant individuals in field populations have provided an early warning of resistance in northern China10,11,12,13. Various mechanisms of resistance to Cry1Ac have been identified previously in H. armigera, including qualitative changes in or reduced levels of the confirmed and putative midgut receptors cadherin, aminopeptidase, alkaline phosphatase and ABCC2 proteins14,15,16,17. In addition, a serine protease from the midgut of a Cry1Ac-resistant strain of H. armigera from India had significantly reduced expression, resulting in improper processing of the protoxin18. Our previous work with ten laboratory-selected strains of H. armigera from China showed that total protease activity in the larval gut was negatively associated with resistance to Cry1Ac19, which suggests that reduced protease activity may contribute to resistance in these strains.
Here we evaluated the role of five trypsin genes in resistance to Cry1Ac in one of the laboratory-selected strains of H. armigera from China. We found that transcription of one of the five genes, which we name HaTryR, was greatly reduced in the resistant strain. We also discovered that silencing of HaTryR with RNA interference (RNAi) increased survival of susceptible larvae exposed to Cry1Ac and that resistance to Cry1Ac was genetically linked with HaTryR. We identified mutations in HaTryR promoter elements associated with resistance that implicate cis-mediated down-regulation of HaTryR transcription as a mechanism of resistance in H. armigera.
Results
Resistance to Cry1Ac protoxin and activated toxin
Bioassay results indicated that, relative to its unselected parent strain (LF), the laboratory-selected LF5 strain of H. armigera was resistant to Cry1Ac protoxin and Cry1Ac activated toxin (Table 1). We calculated the resistance ratios for LF5 as the concentration of Cry1Ac (ng Cry1Ac per g diet) killing 50% of larvae (LC50) for LF5 divided by the LC50 for LF. The resistance ratios were 110 for protoxin and 39 for activated toxin (Table 1).
Abundance of five trypsin mRNAs in midguts of resistant and susceptible larvae
We compared the abundance of five trypsin mRNAs in the midguts of fifth instars between the resistant and susceptible strains. Quantitative real time PCR (qRT-PCR) showed 99% lower abundance of the mRNA for one trypsin gene, HaTryR, in the resistant strain compared to the susceptible strain (P < 0.01, Figure 1). For the other four trypsin genes, no significant difference occurred between the resistant and susceptible strains (Figure 1). These results suggest that decreased expression of HaTryR might contribute to resistance by decreasing the amount of activated toxin in the gut of resistant insects.

Relative abundance of mRNA transcripts of five trypsin genes based on qPCR of resistant and susceptible H. armigera larvae.
Error bars indicate SEM (n = 5 larvae per bar). Asterisks indicate that the expression of HaTryR was significantly lower in resistant larvae than in susceptible larvae (t- test, df = 4, t = −14.3, P < 0.01).
Effects of HaTryR dsRNA on susceptible larvae
For susceptible (LF) larvae, RNA interference (RNAi) by feeding droplets of HaTryR double-stranded RNA (dsRNA) significantly reduced the abundance of HaTryR mRNA in the midgut relative to larvae fed GFP dsRNA as a control (Figure 2A). When fed diet treated with Cry1Ac protoxin, susceptible larvae that had ingested HaTryR dsRNA had twice the survival of larvae that had ingested GFP dsRNA (Figure 2B). These data demonstrate that reducing the expression of HaTryR in susceptible insects decreased their susceptibility to Cry1Ac protoxin.

Effects of HaTryR dsRNA on susceptible H. armigera larvae.
A. Relative abundance of HaTryR mRNA in LF larvae fed GFP dsRNA (control) or HaTryR dsRNA at 1, 2 and 3 days after last ingestion of dsRNA. B. Survival (mean ± SE) of third instar LF larvae fed diet treated with 120 μg Cry1Ac protoxin per gram diet for larvae previously treated with either GFP dsRNA (control) or HaTryR dsRNA. Asterisks indicate significant differences between larvae treated GFP dsRNA and HaTryR dsRNA (t-tests, P < 0.01 in each of the six comparisons).
Mutations in the promoter of the resistant HaTryR allele
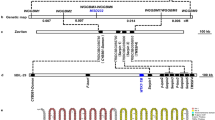
Cloning and sequencing of the genomic DNA of the promoter region of HaTryR in the resistant and susceptible strains revealed several insertions, deletions and substitutions (Figure 3). These differences include four insertions (5 bp from -489 to -485, 1 bp at -447, 4 bp from -407 to -404 and 36 bp from -364 to -329), five deletions (4 bp from -359 to -355, 1 bp at -306, 3 bp from -283 to -281, 29 bp from -227 to -199 and 9 bp from -124 to -116) and 99 substitutions in the promoter region (-515 to -1), as well as two substitutions in the 5′ untranslated region (5′ UTR, +1 to +39) (Figure 3). The 36 bp insertion from -364 to -329 introduces three putative cis-acting regulatory elements: HLF, GATA-1 and CAP (Figure 3). The 29 bp deletion from -227 to -199 removes the putative YY1 element, whereas the 9 bp deletion from -124 to -116 omits the 5′ eight nucleotides of the putative C/EBP element. In addition, the T-392 A-382 substitution may disrupt the putative Ind element (Figure 3).

Alignment of the 5′ flanking sequence of HaTryR from the midgut of Cry1Ac-susceptible (LF) and -resistant (LF5) H. armigera larvae.
The putative cis-acting regulatory elements are shown with lines. Inr+1 indicates the predicted transcription start site (TSS). Start codon (ATG) is marked by a box.
Activity of HaTryR promoters from susceptible and resistant insects in Sf9 cells
To determine if the mutations in the promoter region of the resistance allele reduce transcription of HaTryR, we compared the promoter activity of Sf9 cells transfected with the resistant or susceptible promoter-pYr-PromDetect construct. The activity of the resistant promoter was 7-fold lower than that of the susceptible promoter (p < 0.01, Figure 4). These data suggest that lower expression of HaTryR in the resistant insects was caused by the mutations in the promoter region of the resistant HaTryR allele.

Analysis Cry1Ac-susceptible and resistant HaTryR promoter sequences in transfected Sf9 cells.
Promoter activities were calculated as the activity of sea pansy (Renilla reniformis) luciferase (Rluc) relative to the activity of firefly luciferase (Fluc). Sf9 cells transfected with pYr-PromDetect empty vector were used as a negative control (Control). Means with different letters were significantly different (P < 0.05, n = 3).
Genetic linkage between HaTryR and Cry1A resistance
Sequencing of the HaTryR promoter region in larvae from each of two single-pair backcross families tested in bioassays shows genetic linkage between HaTryR and resistance to Cry1Ac (Table 2). In each of two backcross families (see Methods), the frequency of the putative HaTryR resistance allele was significantly higher than expected at random for larvae that survived exposure to Cry1Ac (Fisher's exact test, P < 0.0001, Table 2). By contrast, in larvae from each backcross family fed untreated diet, the observed frequency of the putative resistance allele did not differ from the 0.50 frequency expected under random assortment (Fisher's exact test, P > 0.60, Table 2).
Discussion
Evaluation of five trypsin genes in H. armigera larvae revealed 99% reduced transcription of one trypsin gene (HaTryR) in the laboratory-selected LF5 strain, which had 110-fold resistance to Cry1Ac protoxin relative to its unselected parent strain (LF). Reducing the expression of this gene with RNAi increased the survival of susceptible larvae exposed to Cry1Ac protoxin. These results indicate that reduced transcription of HaTryR in H. armigera larvae decreased their susceptibility to Cry1Ac. We identified mutations in the promoter region of HaTryR in the resistant strain and found that in Sf9 cells, transcription driven by the resistant HaTryR promoter was lower than transcription driven by the susceptible HaTryR promoter. These results suggest that cis-mediated down regulation of HaTryR causes reduced transcription of this gene and contributes to resistance in the LF5 strain. It remains to be determined which of the several mutations we found in the promoter region contribute to this reduced transcription. As far as we know, this is the first report of cis-mediated down regulation of transcription associated with Bt resistance.
We also found that resistance to Cry1Ac was genetically linked with HaTryR in two backcross families. Because no crossing-over occurs in female Lepidoptera during oogenesis20, the significant association between resistance to Cry1Ac and the HaTryR locus in family A (female F1 X resistant male) indicates that one or more genes conferring resistance are on the same chromosome as HaTryR. Because crossing over does occur in male Lepidoptera during spermatogenesis20, the significant association between resistance to Cry1Ac and the HaTryR locus in family B (male F1 X resistant female) indicates that one or more genes conferring resistance are at or near the HaTryR locus. If the gene(s) conferring resistance are on the same chromosome as HaTryR, but not at the same locus, a stronger association is expected between resistance and the putative HaTryR allele in family A (no crossing-over) than in family B (crossing-over). This association, however, was not significantly stronger in family A (101 rr: 19 rs) than in family B (96 rr: 24 rs) (P = 0.50), which implies that resistance is tightly linked with the HaTryR locus. The finding that some rs survived on treated diet (19 larvae in family A and 24 larvae in family B) indicates that inheritance of resistance conferred by the HaTryR resistance allele is not completely recessive, one or more genes not linked with HaTryR contributed to resistance, or both.
The results here showing an association between reduced transcription of a trypsin gene and resistance to Cry1Ac in the laboratory-selected LF5 strain of H. armigera from China are similar to previous work showing an association between reduced transcription of a serine protease gene (HaSP2) and resistance to Cry1Ac in a laboratory-selected strain of H. armigera from India18. Reduced transcription of HaSP2 was associated with reduced activation of Cry1Ac protoxin18. The resistance ratio for the laboratory-selected Indian strain was 72 for Cry1Ac protoxin, but only 1.2 for Cry1Ac activated toxin, implying that reduced activation of Cry1Ac was responsible for all or nearly all of the resistance18. By contrast, the resistance ratio for LF5 was 110 for Cry1Ac protoxin and 39 for Cry1Ac activated toxin, indicating that a mechanism other than reduced activation of Cry1Ac contributes substantially to resistance in LF5.
Protease-mediated resistance was first demonstrated in a laboratory-selected strain of Plodia interpunctella selected for resistance to Bt subspecies entomocidus6. Reduction of protease activity has also been associated with resistance to Cry1A toxins in laboratory-selected strains of Heliothis virescens21 and Ostrinia nubilalis22,23. In one resistant strain of O. nubilalis, the resistance ratio was 250 for Cry1Ab protoxin compared with 12 for Cry1Ab activated toxin23. In a field-selected resistant strain of Plutella xylostella from Malaysia, a substrain that was not exposed to Cry1Ac in the laboratory had a resistance ratio of 315 to Cry1Ac protoxin compared with 25 for activated toxin24. Selection with activated Cry1Ac in the laboratory boosted the resistance ratios to >15,000 for Cry1Ac protoxin versus 94 for Cry1Ac activated toxin24.
In conjunction with results from several previous studies, the new data reported here show that in some cases, pest resistance is lower to Bt activated toxin than protoxin (but see25 for a notable exception). In such cases, transgenic plants that produce activated toxins could be more durable than plants that produce protoxins or partially activated toxins. Implementing this idea in China would favor planting of the GK type of Bt cotton producing truncated Cry1Ac rather than Bt cotton that produces full-length Cry1Ac26,27. However, resistance management strategies for H. armigera and other pests must also consider mechanisms that confer resistance to activated toxins.
Methods
Insects and resistance selection
We used a Cry1Ac-susceptible strain of H. armigera (LF), which was started with insects collected from Langfang, Hebei Province, China in 1998 and had been reared in the laboratory for >15 years on artificial diet without exposure to Bt toxins19. We generated the LF5 strain by selecting the LF strain initially at 1 µg Cry1Ac protoxin per g diet for 38 generations. We then increased the selection concentration for LF5 to 5 µg Cry1Ac protoxin per g diet for 104 generations of selection.
Insect bioassays
Cry1Ac protoxin and activated toxin were kindly supplied by the Biotechnology Research Group, Institute of Plant Protection, Chinese Academy of Agricultural Science. For activation, Cry1Ac protoxin was incubated 2 h at 37°C with 1/25 ratio of bovine pancreas trypsin (Sigma) and the soluble trypsinized toxin was purified by a Superdex 200 HR 10/30 column (Amersham Biosciences) on a fast protein liquid chromatography (FPLC) system. H. armigera bioassays with Cry1Ac protoxin and activated toxin were performed with a diet incorporation procedure. Approximately 1–1.5 g artificial diets incorporated with various concentrations of Cry1Ac protoxin or toxin suspended in distilled water or equal volume of distilled water (control) was put in each well of 24-well plates. A single first instar larva was placed into each well and 24 larvae were used for each treatment. Mortality was recorded after seven days and all assays were replicated three times. Pooled data were subjected to statistical analysis; the concentration killing 50% of larvae (LC50) was calculated by probit analysis with the computer program POLO (LeOra Software, Berkeley, California 1987).
Quantitative RT-PCR
Sequences encoding trypsin genes from H. armigera were retrieved from GenBank and were used compare gene expression in the Cry1Ac-susceptible and resistant strains: HaTry1 (EU98284128), HaTry2 (EU32554929), HaTry3 (EU32554829), HaTry4 (AY437836) and HaTryR (KF791044). Total RNA was extracted from the midgut of fifth instars of susceptible or resistant strains with TRIzol® reagent (Invitrogen) according to the manufacturer's instructions and was treated with DNase I (Takara) to remove any residual DNA and was reverse-transcribed with SuperScript III RNase H− reverse transcriptase (Invitrogen).
Quantitative real-time PCR (qPCR) amplification and analysis were carried out using the Applied Biosystems 7500 Fast Real-time PCR System. The transcriptional profiles of selected H. armigera trypsin genes were analyzed using the SYBR Premix Ex TaqTM (TaKaRa) system. Gene-specific primers designed for real-time PCR are listed in Table 3. H. armigera actin (GenBank accession no. X97615) and GADPH (GenBank accession no. JF417983) were used as controls. The PCR involved a 95°C step for 30 s followed by 40 cycles at 95°C for 5 s, 58°C for 15 s and 68°C for 30 s. All reactions were run in triplicate with three independent biological replicates and monitoring the dissociation curve to control potential formation of primer dimers. PCR efficiency was determined by a series of 3-fold dilutions of cDNA from LF larvae. The absolute value of the slope (Ct value Vs Log) for each primer set was <0.1 and all amplification efficiencies were >99% when compared to the endogenous control. Data obtained were analyzed using the relative 2-ΔΔCT quantitation method to calculate transcript abundance, which was standardized to 1 for reactions with samples from susceptible larvae. We calculated percentage reduction in mRNA as the relative abundance of mRNA in susceptible larvae (1) minus the relative abundance in resistant larvae multiplied by 100%.
Cloning and sequence analysis of HaTryR
The 3′- rapid amplification of cDNA ends (RACE) and 5′-RACE were performed using 3′-full RACE Core Set Ver. 2.0 Kit and 5′-full RACE Kit (TaKaRa) to obtain the cDNA 3′ and 5′ ends of the H. armigera trypsin gene HaTryR (primers are listed in Table 3). All the amplified PCR products were recovered and cloned into the pMD20-T Vector (TaKaRa) for sequencing.
The NCBI/BLAST database and ClustalX30 were used to analyse the homology of H. armigera trypsin genes. Multiple alignments and identity calculations were done with DNAMAN 6.0 (Lynnon Corporation).
RNAi-mediated gene silencing and survival assays
Primers were designed for HaTryR and green fluorescent protein (GFP) genes and the T7 primer sequence was added to both forward and reverse primers (Table 3). The dsRNA for RNAi was prepared using PCR products as a template for in vitro transcription. In vitro transcription to yield dsRNA of HaTryR was performed with T7 RNA polymerase using the HiScribe RNAiTM T7 In Vitro Transcription Kit (New England Biolabs) according to the manufacturer.
In terms of dsRNA delivery to third instar of H. armigera, oral delivery was more suitable for our purpose. The technique is not invasive (no mortality) and the larvae were immediately subjected to subsequent toxicity analysis. The 3rd instar larvae of LF were individually placed into each well of 24-well plates to avoid cannibalism and starved for 12 h. Seventy-two larvae were then fed with a 2 µl drop of 2.5 µg dsRNA from either HaTryR or GFP (nonspecific dsRNA as control) respectively in delivery buffer (10 mM Tris-Cl, pH 7.5; 10 mM EDTA). After 2 h, droplet-fed larvae were placed back into a well of a 24-well plate provided of artificial diet. Oral delivery was repeated three more times, with a total of 10 µg dsRNA fed over a 2 d interval. After the 2nd feeding of dsRNA, droplet-fed larvae were placed back into a well of a 24-well plate containing artificial diet that either 120 µg/g Cry1Ac protoxin or no treatment. Survival was assessed after 0, 3, 6 and 9 days. The experiment was repeated twice. To evaluate knockdown, we checked the expression levels of HaTryR by RT-PCR (1, 2, 3 d) after the last oral delivery by qRT-PCR as described above.
Promoter cloning and sequences analysis
Genomic DNA of susceptible and resistant strains of H. armigera was extracted using Tiangen Genomic DNA kit (TianGen Biotech Co., Ltd, Beijing). Nested specific primers (SP1, SP2, SP3) were designed to the HaTryR genomic DNA sequence (Table 3). Arbitrary primers (AP1, AP2, AP3) were provided with the Universal Genome Walking kit (TaKaRa). Three-rounds of TAIL-PCR were performed according to the manufacturer's instructions. After TAIL-PCR, DNA products were amplified from AP3 and SP3 primers and were cloned into the PMD 20-T vector (TaKaRa). The cloned plasmids were sequenced and similarity analysis and alignment were by Sequencher (version 4.1.4; Gene Codes Corporation). The TRANSFAC database (v. 1.3; http://www.cbrc.jp/research/db/TFSEARCH.html) was used to search for transcription factor binding sites. A search for promoter elements was performed with TESS (http://www.cbil.upenn.edu/cgi-bin/tess/tess). The sequence upstream of the putative transcriptional start site was predicted by the Neural Network Promoter Prediction website (http://www.fruitfly.org/seq_tools/promoter.html).
Transient transfections and dual luciferase assays
We constructed two reporter plasmids encompassing −515 to +39 bp (545 bp) of the susceptible HaTryR promoter and −515 to +39 bp (545 bp) of the resistant promoter by PCR of genomic DNA from susceptible and resistant strains, respectively, using the primers in Table 3. Amplicons were cloned into the pMD-19T simple vector (Takara) following the manufacturer's instructions. The recombinant plasmid was excised with Bgl II and Nhe I and ligated into a pYr-PromDetect vector harboring dual reporters to improve accuracy (YRBIO, China). We used simultaneous measurement of two individual reporter enzymes within a single construct to evaluate promoter activity31. Briefly, the activity of the “experimental” reporter (luciferase from the sea pansy, Renilla reniformis) is correlated with the effect of the specific promoter, while the activity of the “control” reporter (firefly luciferase) with an HSV-TK promoter provides an internal control that serves as the baseline response. All constructs used in this study were restriction mapped and sequenced to confirm their authenticity.
We seeded 1.5 × 105 Sf9 cells per well in 24-well plates and transfected with pYr-PromDetect constructed without or with a susceptible or resistant HaTryR promoter using Lipofectamine 2000 (Invitrogen) and Opti-MEM (Gibco). All transfections were carried out in triplicate. After 48 h incubation, we collected cells and calculated promoter activity as the activity of sea pansy luciferase relative to the activity of firefly luciferase.
Genetic Linkage Analysis
For linkage analysis of the resistant HaTryR promoter and Cry1Ac resistance, a single-pair cross was obtained from mating a susceptible male and resistant female for F1 progeny and two backcross families were from a single-pair cross of female F1 progeny with resistant male (Backcross family A) and resistant female (Backcross family B). Backcross families were reared on an artificial diet (non-Cry1Ac-selected) or treated with 5 μg/g of Cry1Ac protoxin for 10 days to select Cry1Ac-resistant individuals (Cry1Ac-selected) as described elsewhere32. Larval genomic DNA was prepared from individual larvae and a 600-bp genomic DNA fragment including the HaTryR promoter was amplified by PCR and sequenced to determine the allele types (primers are listed in Table 3) and the results were analyzed by using χ2 test.
References
Mendelsohn, M., Kough, J., Vaituzis, Z. & Matthews, K. Are Bt crops safe? Nat Biotechnol 21, 1003–1009 (2003).
Pardo-López, L., Soberón, M. & Bravo, A. Bacillus thuringiensis insecticidal three-domain Cry toxins: mode of action, insect resistance and consequences for crop protection. FEMS Microbiol Rev 37, 3–22 (2013).
James, C. Global Status of Commercialized Biotech/GM Crops. ISAAA Briefs 46, (ISAAA, Ithaca, NY, 2013) (2013).
Tabashnik, B. E., Brévault, T. & Carrière, Y. Insect resistance to Bt crops: lessons from the first billion acres. Nat Biotechnol 31, 510–521 (2013).
Ferré, J. & van Rie, J. Biochemistry and genetics of insect resistance to Bacillus thuringiensis. Annu Rev Entomol 47, 501–533 (2002).
Oppert, B., Kramer, K. J., Beeman, R. W., Johnson, D. & McGaughey, W. H. Proteinase-mediated insect resistance to Bacillus thuringiensis toxins. J Biol Chem 272, 23473–23476 (1997).
Wu, Y. Detection and mechanisms of resistance evolved in insects to Cry toxins from Bacillus thuringiensis. Adv Insect Physiol 2014 (in press).
Wu, K. & Guo, Y. The evolution of cotton pest management practices in China. Annu Rev Entomol 50, 31–52 (2005).
Wu, K., Lu, Y., Feng, H., Jiang, Y. & Zhao J. Suppression of cotton bollworm in multiple crops in China in areas with Bt toxin-containing cotton. Science 321, 1676–1678 (2008).
Akhurst, R. J., James, W., Bird, L. J. & Beard, C. Resistance to the Cry1Ac δ-endotoxin of Bacillus thuringiensis in the cotton bollworm, Helicoverpa armigera (Lepidoptera: Noctuidae). J Econ Entomol 96, 1290–1299 (2003).
Xu, X., Yu, L. & Wu, Y. Disruption of a cadherin gene associated with resistance to Cry1Ac δ-endotoxin of Bacillus thuringiensis in Helicoverpa armigera. Appl Environ Microbiol 71, 948–954 (2005).
Kranthi, K. R. et al. Inheritance of resistance in Indian Helicoverpa armigera (Hübner) to Cry1Ac toxin of Bacillus thuringiensis. Crop Prot 25, 119–124 (2006).
Luo, S. et al. Binding of three Cry1A toxins in resistant and susceptible strains of cotton bollworm (Helicoverpa armigera). Pestic Biochem Physiol 85, 104–109 (2006).
Zhang, H. et al. Diverse genetic basis of field-evolved resistance to Bt cotton in cotton bollworm from China. Proc Natl Acad Sci USA 109, 10275–10280 (2012).
Zhang, S. et al. Mutation of an aminopeptidase N gene is associated with Helicoverpa armigera resistance to Bacillus thuringiensis Cry1Ac toxin. Insect Biochem Mol Biol 39, 421–429 (2009).
Jurat-Fuentes, J. L. et al. Reduced levels of membrane-bound alkaline phosphatase are common to Lepidopteran strains resistant to Cry toxin from Bacillus thuringiensis. PLoS ONE 6, e17606 (2011).
Xiao, Y. et al. Mis-splicing of the ABCC2 gene linked with Bt toxin resistance in Helicoverpa armigera. Sci Rep 4, 6184 (2014).
Rajagopal, R. et al. Resistance of Helicoverpa armigera to Cry1Ac toxin from Bacillus thuringiensis is due to improper processing of the protoxin. Biochem J 419, 309–316 (2009).
Cao, G., Zhang, L., Liang, G., Li, X. & Wu, K. Involvement of nonbinding site proteinases in the development of resistance of Helicoverpa armigera (Lepidoptera: Noctuidae) to Cry1Ac. J Econ Entomol 106, 2514–2521 (2013).
Heckel, D. G., Gahan, L. J., Liu, Y. B. & Tabashnik, B. E. Genetic mapping of resistance to Bacillus thuringiensis toxins in diamondback moth using biphasic linkage analysis. Proc Natl Acad Sci USA 96, 8373–8377 (1999).
Karumbaiah, L., Oppert, B., Jurat-Fuentes, J. L. & Adang, M. J. Analysis of midgut proteinases from Bacillus thuringiensis-susceptible and -resistant Heliothis virescens (Lepidoptera: Noctuidae). Comp Biochem Physiol B Biochem Mol Biol 146, 139–146 (2007).
Li, H. et al. Comparative analysis of proteinase activities of Bacillus thuringiensis-resistant and -susceptible Ostrinia nubilalis (Lepidoptera: Crambidae). Insect Biochem Mol Biol 34, 753–762 (2004).
Li, H. et al. Characterization of cDNAs encoding three trypsin-like proteinases and mRNA quantitative analysis in Bt-resistant and -susceptible strains of Ostrinia nubilalis. Insect Biochem Mol Biol 35, 847–860 (2005).
Sayyed, A. H., Gatsi, R., Kouskoura, T., Wright, D. J. & Crickmore, N. Susceptibility of a field-derived, Bacillus thuringiensis-resistant strain of diamondback moth to in vitro-activated Cry1Ac toxin. Appl Environ Microbiol 67, 4372–4373 (2001).
Caccia, S. et al. Association of Cry1Ac toxin resistance in Helicoverpa zea (Boddie) with increased alkaline phosphatase levels in the midgut lumen. Appl Environ Microbiol 78, 5690–5698 (2012).
Wang, Y., Xie, J., Zhang, Y., Wang, X. & Peng, Y. A PCR method to detect different Bt gene expression cassettes in transgenic Bt cotton. J Agric Biotechnol 17, 914–919 (2009).
Yu, S. & Fan, S. Research and commercialization of national Bt cotton in China. Biobusiness 3, 35–41 (2010).
Liu, Y., Sui, Y., Wang, J. & Zhao, X. Characterization of the trypsin-like protease (Ha-TLP2) constitutively expressed in the integument of the cotton bollworm, Helicoverpa armigera. Arch Insect Biochem Physiol 72, 74–87 (2009).
Campbell, P. M., Cao, A. T., Hines, E. R., East, P. D. & Gordon, K. H. J. Proteomic analysis of the peritrophic matrix from the gut of the caterpillar, Helicoverpa armigera. Insect Biochem Mol Biol 38, 950–958 (2008).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25, 4876–4882 (1997).
Wood, K. V. The chemistry of bioluminescent reporter assays. Promega Notes 65, 14 (1998).
Tiewsiri, K. & Wang, P. Differential alteration of two aminopeptidases N associated with resistance to Bacillus thuringiensis toxin Cry1Ac in cabbage looper. Proc Natl Acad Sci USA 108, 14037–14042 (2011).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 31210103921, No. 31201525 and No. 31321004). Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Contributions
C.L. and K.W. designed the experiments; C.L. and Y.X. performed the experiments; C.L., Y.X., X.L., B.E.T., B.O. and K.W. performed the statistical analysis; and C.L., X.L., B.O., B.E.T. and K.W. wrote the manuscript. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare competing interests. B.E.T. is coauthor of a patent on modified Bt toxins, "Suppression of Resistance in Insects to Bacillus thuringiensis Cry Toxins, Using Toxins that do not Require the Cadherin Receptor" (patent numbers: CA2690188A1, CN101730712A, EP2184293A2,EP2184293A4, EP2184293B1, WO2008150150A2, WO2008150150A3). Pioneer, Dow AgroSciences, Monsanto and Bayer CropScience did not provide funding to support this work, but they have funded other work by B.E.T.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Liu, C., Xiao, Y., Li, X. et al. Cis-mediated down-regulation of a trypsin gene associated with Bt resistance in cotton bollworm. Sci Rep 4, 7219 (2014). https://doi.org/10.1038/srep07219
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07219
This article is cited by
-
Diversity of transgenes in sustainable management of insect pests
Transgenic Research (2023)
-
Key residues of Bacillus thuringiensis Cry2Ab for oligomerization and pore-formation activity
AMB Express (2021)
-
Alterations in the expression of certain midgut genes of Spodoptera littoralis (Boisd.) (Lepidoptera: Noctuidae) larvae and midgut histopathology in response to Bacillus thuringiensis Cry1C toxin
Egyptian Journal of Biological Pest Control (2021)
-
Insect resistance management in Bacillus thuringiensis cotton by MGPS (multiple genes pyramiding and silencing)
Journal of Cotton Research (2020)
-
Differences in midgut transcriptomes between resistant and susceptible strains of Chilo suppressalis to Cry1C toxin
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
