
Abstract
The liver is unique in its capacity to regenerate after injury, during which hepatocytes actively divide and establish cell-cell contacts through cell adhesion complexes. Here, we demonstrate that the loss of α-catenin, a well-established adhesion component, dramatically disrupts liver regeneration. Using a partial hepatectomy model, we show that regenerated livers from α-catenin knockdown mice are grossly larger than control regenerated livers, with an increase in cell size and proliferation. This increased proliferation correlated with increased YAP activation, implicating α-catenin in the Hippo/YAP pathway. Additionally, α-catenin knockdown mice exhibited a phenotype reminiscent of clinical cholestasis, with drastically altered bile canaliculi, elevated levels of bile components and signs of jaundice and inflammation. The disrupted regenerative capacity is a result of actin cytoskeletal disorganisation, leading to a loss of apical microvilli, dilated lumens in the bile canaliculi and leaky tight junctions. This study illuminates a novel, essential role for α-catenin in liver regeneration.
Similar content being viewed by others


Introduction
Liver diseases are prevalent worldwide, with American statistics reporting that one in ten people suffer from some form of liver disease, such as fatty liver, hepatitis C or liver cancer1. Liver disease is also reported to be the European Union's fifth leading cause of death, accounting for at least one in six deaths2. Although some specific therapies are available to treat liver diseases, survival predominantly hinges on the regenerative potential of the liver. Unlike other organs, the liver is unique in its capacity to recover from injury by regeneration instead of scar formation3. In light of this, liver regeneration has been studied extensively over the past few decades in an effort to translate treatment strategies into the clinic. The best-characterised paradigm for investigating liver regeneration has been through the use of partial hepatectomy in rodents, which results in a liver-specific regenerative response4. During liver regeneration, different intracellular and extracellular signals are stimulated, which in turn activate quiescent hepatocytes to re-enter the cell cycle and proliferate, restoring the liver-to-body mass ratio. The ensuing remodelling of the liver architecture accommodates this increase in cell number and restores the liver to its normal state prior to regeneration5.
During liver regeneration, newly formed hepatocytes organise into highly polarised cords through the assembly of junctional complexes. Adherens junctions, which are the first critical event in junction formation, initiate from a cadherin-catenin complex. Classical cadherins are transmembrane glycoproteins that ensure intercellular adhesion through homophilic interactions and are indirectly connected to the cortical actin cytoskeleton through α-catenin. Alpha-catenin is a mechanosensor and mechanotransducer with two actin-binding domains6. The carboxy-terminal domain can either bind directly to actin microfilaments (if α-catenin is subjected to force-induced trans-conformation)7,8,9 or to EPLIN (epithelial protein lost in neoplasm)10, which serves as an actin-bundling filament. The central domain, which needs to be unmasked through acto-myosin force generation at the C-terminus, becomes accessible for vinculin binding, which, in turn, promotes actin microfilament assembly6. The adhesive strength is thus maximised when the two sites act in concert11, anchoring the actin filaments through both EPLIN and vinculin to modulate cytoskeletal changes in response to extracellular signals12.
Several studies have recently emerged demonstrating new roles for α-catenin. Rather than just a linker protein, α-catenin is reported to act as a molecular switch in the control of actin cytoskeletal organisation during adhesion7,13. In addition, ablation of α-catenin in the mouse epidermis results in defects reminiscent of squamous cell carcinoma, including hyper-proliferation and irregularities in cell polarity14. This anti-proliferative role of α-catenin has since been substantiated with the use of conditional α-catenin knockout mice targeting the brain15, as well as in studies that have connected α-catenin with the Hippo pathway16. Also, a role for α-catenin in the modulation of microtubules has been highlighted17.
Previous work has debated whether proliferation in the liver is attributed to liver stem cells or adult hepatocytes, but a conclusive study has elucidated that 99% of new hepatocytes arise from pre-existing adult hepatocytes18. Hence, examination of the effect of α-catenin knockdown in existing hepatocytes and how this affects proliferation and the formation of new cell-cell contacts may shed light on the role of α-catenin in the process of liver regeneration.
Results
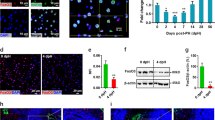
To determine the in vivo significance of α-catenin in liver regeneration, a liver-specific knockdown of this gene was accomplished through α-catenin siRNA packaged into lipid nanoparticles (LNPs) and modified to specifically target the liver19,20,21,22. Livers and kidneys as a control organ, were obtained from mice injected with the siRNA-LNP on an extended regime (Fig. S1A). These tissues were then analysed for transcript levels to ensure knockdown and specificity. A significant reduction in α-catenin expression was observed in the α-catenin siRNA-injected livers as compared with control siRNA-injected livers, whereas the control organ (kidney) did not show any change in transcript levels between the two groups (Fig. 1A). Similarly, we observed a corresponding reduction in protein levels, as determined by western blotting and immunohistofluorescence (Fig. 1B, Fig. S1B–C), the latter showing that α-catenin was uniformly silenced throughout the tissue, predominantly in the hepatocytes.

Specific knockdown of α-catenin in liver induces major alterations to the hepatic functional unit.
(A) α-catenin siRNA-nanolipid particle (LNP)-injected mice exhibited a specific knockdown in α-catenin transcript levels (n = 6 for each group) in liver but not in kidney (n = 4 for each group). (B) α-catenin protein levels in liver showed a corresponding decrease (n = 6 for each group). Gel image shown is cropped for concise presentation. (C) Regenerated liver tissues were immunolabelled with antibodies directed against collagen IV (red; 10× and 20×) and zona occludens (ZO)-1 (green; 40× magnification). Sinusoids labelled with collagen IV were tortuous and disorganised in α-catenin siRNA-treated livers. Bile canaliculi immunolabelled with ZO-1 were arranged in an organised network of tubular structures, whereas a disruption in this network was observed in α-catenin siRNA-injected livers. The lumens of the BC were also enlarged. (D) Upon harvesting, the livers were dissected and weighed. The α-catenin siRNA-injected livers were 50% bigger than the control livers. (E) Immunolabelling of cell membranes of liver sections with N-cadherin (red; 20× magnification) revealed that some cells in α-catenin siRNA-injected livers were significantly larger. (F) A statistical analysis of the cell size profile of the experimental livers was performed. Top: The probability distribution graph of the cells was computed and shown. Bottom: The difference between control and α-catenin siRNA cell populations was computed and is represented by the percentage change between the siRNA and control populations. This shows that α-catenin siRNA-injected livers were more highly enriched in the smaller and larger cell fractions as compared with control livers (Control: n = 8, α-cat: n = 11). Scale bars, 50 µm.
The liver is made up of functional units consisting of hepatocyte cords, which form bile canaliculi and the blood vessels between these cords. The vascular network forms sinusoids, which are orderly, well-spaced tubular vessels lining the basolateral membranes of hepatocytes. On the apical membrane, bile canaliculi are lined up in a well-organised network of tubules sealed by tight junctions. The polarisation of the hepatocyte plasma membrane is critical for the localisation of transporters, which are essential for hepatocyte function. To investigate the role of α-catenin in the structural formation and maintenance of the liver, a well-established liver regeneration model (partial hepatectomy) was chosen for its reproducibility and efficacy4 (Fig. S1A). Under surgical conditions, two-thirds of the liver was removed and the regenerated lobes were harvested seven days later; this time period is sufficient to allow for more than 70% regeneration of the liver mass4. Interestingly, we found that α-catenin knockdown livers exhibited a vastly perturbed phenotype. There was a disruption of hepatic cords associated with hepatocellular hypertrophy and hyperplasia in knockdown mice. The hypertrophic hepatocytes were enlarged with granular or “ground glass” deep eosinophilic cytoplasms and enlarged nuclei, predominantly observed in periportal and mid-zonal areas and, to a lesser extent, around the central vein (Fig. S2). The sinusoids in knockdown livers were highly tortuous, disorganised and unevenly spaced. The bile canalicular network, as visualized by ZO-1 immunolabeling, was also grossly disorganised, with the normally branched network replaced with misshapen, disconnected structures with enlarged lumens (Fig. 1C). In order to ascertain that this disorganization was attributed to α-catenin knockdown in hepatocytes specifically, we silenced α-catenin in AML12 cells, a mouse hepatocyte cell line known to exhibit typical hepatocyte features such as peroxisomes and bile canalicular-like structures. The knockdown of α-catenin elicited a disruption in ZO-1 labelling reminiscent of that observed in vivo (Fig. S3A). Similarly, when α-catenin was silenced in isolated primary hepatocytes, the bile canaliculi exhibited enlarged lumens, as observed in vivo (Fig. S3B). We also noted hepatomegaly in the knockdown livers, with enlargements at both the physical and cellular levels and a 60% increase in the liver-to-body weight ratio (Fig. 1D). Upon closer inspection, we saw that the knockdown livers possessed a greater percentage of larger hepatocytes (Fig. 1E). Further analysis of cell size showed a great variation in cell size in the knockdown livers, with a significantly greater percentage of cells that were smaller and larger than the median size (100–250 µm), which led to a corresponding decrease in the median size as compared with the controls (Fig. 1F). This non-uniformity in hepatocyte cell size reflects a general disorganisation of the liver, as hepatocytes are usually uniform in size so as to maintain the ordered arrangement of vessels along the hepatocyte cords. At the morphological level, our results indicate that the knockdown of α-catenin impaired the mechanisms of liver regeneration to induce a severely disorganised liver structure.
To further investigate the effect of this disorganisation on liver metabolism, several liver enzymes were measured from blood samples obtained from these mice. Interestingly, α-catenin knockdown mice exhibited a significant increase in alkaline phosphatase (ALP) and total bilirubin (TBIL) levels. Clinically, these changes are indicative of a disruption in bile flow, which manifests as a medical condition known as cholestasis in humans (Fig. 2A). Amylase (AMY) levels were also elevated, which may occur following the blockage of bile transport (Fig. 2A). A subset of mice also exhibited signs of jaundice, with yellow skin pigmentation, which is consistent with the elevated bilirubin levels (data not shown). Since a disruption in bile flow would imply an accumulation of bile acids in the blood circulation, we next evaluated the total bile acid (TBA) levels in the plasma. As expected, we found a 10-fold increase in serum TBA levels in α-catenin knockdown mice as compared with control mice, consistent with a cholestasis-like phenotype (Fig. 2B). In addition, because bile is usually secreted into the stomach for the reabsorption of lipids, a decrease in lipid content would be expected if there were an obstruction in bile flow. Indeed, the level of high density lipoprotein (HDL) was significantly lower in the α-catenin knockdown mice (Fig. 2C). Finally, a haematological analysis was performed to assess for signs of inflammation. The total white blood cell count, including all cellular subtypes (neutrophil, lymphocytes, monocytes and eosinophils), was elevated in the knockdown mice, consistent with an inflammatory response (Fig. 2D).

Blood chemistry of α-catenin-knockdown mice is indicative of cholestasis in vivo.
Blood chemistry analysis was performed on blood samples taken from mice at the time of liver harvesting and several markers linked to liver function were measured. (A) Alkaline phosphatase (ALP) levels were significantly higher in the α-catenin siRNA-injected group. High ALP levels are clinically linked to bile canaliculi malfunction. Total bilirubin (TBIL) content was also elevated in the siRNA-injected group as compared with the control, which suggests a dysfunction in bile production and flow. Amylase (AMY) levels were also significantly higher in the α-catenin-injected group, which is clinically linked to disrupted bile flow (Control: n = 17, α-cat: n = 21). (B) Total bile acid (TBA) levels were also elevated in the α-catenin-injected group, indicating that there is a high build-up of TBA in the blood. (C) High-density lipoprotein (HDL) levels were significantly decreased in α-catenin-injected group, corresponding to a blockage in bile transport (con: n = 9, α-cat: n = 10). (D) siRNA-injected mice exhibited a significantly higher total white blood cells (WBC), suggesting inflammation. Furthermore, the different subsets within the white blood cell population were all significantly higher, including neutrophils (NE) and monocytes (MO), lymphocytes (LY) and eosinophils (EO). K/µl: Thousands per microliter of blood (con: n = 17, siRNA: n = 21).
In order to determine if the effect of α-catenin knockdown was pertinent to the regenerative process, α-catenin expression was silenced in the absence of partial hepatectomy. A significantly more modest phenotype was observed. The bile canaliculi exhibited a partial loss of its branched structure with slightly enlarged lumens (Fig. S4A). Alkaline phosphatase and amylase levels were significantly elevated in α-catenin knockdown mice (Fig. S4B–C), but we measured no significant change in total bilirubin levels (Fig. S4D). In addition, there was no significant increase in liver mass in α-catenin knockdown mice (Fig. S4E).
Next, we sought to understand how the loss of α-catenin induced these defects. The observed increase in liver size could be due to an increase in hepatocyte cell size or an increase in cell number. Thus, we generated a liver simulation model to calculate the hypothetical increase in liver size that could be caused solely by an increase in cell size. This analysis indicated that the observed increase in cell size could account for only a 1.3% increase in liver mass (Fig. S5). Thus, to ascertain if the increase in liver size was instead due to proliferation, regenerated liver sections were immunolabelled with the proliferative marker Ki67. Knockdown liver sections showed a much greater extent of positive Ki67 staining as compared with control sections. Quantification of the percentage of Ki67-positive cells showed a 6-fold increase in Ki67-labelled cells in α-catenin knockdown livers as compared with the controls (13.37% versus 2.1%, respectively) (Fig. 3A).

Increase in liver size is caused by an increase in hepatocyte proliferation via the activation of YAP.
(A) Nuclei of cells from liver sections were immunolabelled with anti-Ki67 antibody (green) and DAPI (blue). Left: Representative images of control and α-catenin knockdown liver sections. Scale bars, 50 µm. Right graph: The percentage of Ki67-positive cells out of all nuclei-positive cells was then calculated for α-catenin knockdown and control livers. α-catenin knockdown livers exhibited a much greater level of proliferation (%) as compared with the control livers (13.37% versus 2.1%, respectively). (B) Liver lysates were blotted for phosphorylated (p) YAP and total (t) YAP and the ratio of phosphorylated YAP to total YAP (as a percentage) was significantly lower in the α-catenin knockdown livers (n = 9 for each group). Gel images shown are cropped and representative of gels run under the same experimental conditions. (C) Nuclei of cells from liver sections were immunolabelled with anti-YAP antibody (brown). Top: Representative images of control and α-catenin knockdown liver sections. Scale bars, 50 µm. Left bottom graph: The percentage of YAP-positive (+ve) nuclei was calculated for α-catenin knockdown and control livers (n = 7 for each group). (D) Connective tissue growth factor (CTGF), an important downstream transcriptional target of YAP, was assessed for gene expression levels. CTGF was found to be substantially up-regulated in alpha-catenin knockdown liver (n = 7 for control group, n = 10 for alpha-catenin group).
Previous reports have linked increased proliferation in the liver to the Hippo-YAP pathway. In addition, α-catenin has been identified as a physical interactor of Yap1, a prominent player in the Hippo pathway16. To explore this possible link, we performed western blotting to determine any change in YAP phosphorylation, a post-translational modification that interferes with its nuclear localisation. Interestingly, α-catenin knockdown resulted in a consistent decrease in the percentage of phosphorylated YAP (as a function of total YAP), hence supporting the involvement of this pathway in cell proliferation (Fig. 3B). Given that phosphorylated/activated YAP translocates to the nucleus to activate downstream transcriptional targets, we next sought to determine the percentage of YAP nuclear translocation by quantifying the percentage of YAP-positive nuclei. Alpha-catenin knockdown livers exhibited almost a doubling of the percentage of YAP-positive nuclei, verifying that YAP signalling is indeed potentiated by the loss of α-catenin (Fig. 3C). The decrease in percentage of phosphorylated YAP to total YAP is a decrease from 52% to 32%, which is approximately a 40% decrease. The increase in percentage of YAP-positive nuclei is from 37% to 78%, which is a little over a two-fold increase. Considering that these two results were obtained from different types of assays, we think that the results are strikingly consistent, taking in account an expected level of disparity due to the different nature of the assays. In addition, we examined the expression level of connective tissue growth factor (CTGF), an important YAP downstream target (Fig. 3D). We found that CTGF was significantly up-regulated in α-catenin knockdown liver, substantiating the involvement of the YAP signalling pathway.
In clinical cases of cholestasis, the dysfunction in bile flow observed in patients has been linked to either a change in the expression levels of crucial bile transporters or to cytoskeletal changes, which, in turn, lead to leaky tight junctions that fail to adequately seal the bile canaliculi23. Although the disruption in the bile canalicular network in this case points to the latter theory, we still examined the expression of four different transporters implicated in cholestasis to rule out the possibility24: Multidrug resistance-associated protein-2 (Mrp2), Mrp3, multidrug resistance gene-1 (Mdr1, otherwise known as P-glycoprotein multidrug transporter (Pgp) or ABCB1) and Mdr2. As expected, there were no significant changes in the expression levels of these proteins, suggesting that the disruption of bile flow did not involve changes in the bile transporter proteins (Fig. 4A). Most human and animal cholestatic liver disorders are associated with changes in the hepatocyte cytoskeleton, which includes an accumulation of disorganised actin filament bundles in the pericanalicular domain. This, in turn, results in the loss of apical microvilli and diminished contractility of the canalicular membrane, which may contribute to the leakiness of tight junctions24. Alpha-catenin is known to associate with cortical actin filaments; therefore, we investigated the localisation of actin filaments using phalloidin staining. In the control livers, actin bundles outlined the periphery of the cell membranes in an organised, pericanalicular pattern, whereas, in knockdown livers, the actin filaments were grossly distorted, misshapen and did not continuously line the cell cortex (Fig. 4B).

Bile canaliculi (BC) disruption caused by actin cytoskeleton disorganisation results in a loss of microvilli and dilated BC lumens.
(A) The real-time expression of four important bile transporter proteins— Multidrug resistance-associated protein-2 (Mrp2), Mrp3, multidrug resistance gene-1 (Mdr1, otherwise known as P-glycoprotein multidrug transporter (Pgp) or ABCB1) and Mdr2—was measured in α-catenin knockdown livers and compared with control livers. No significant change was observed for any of the proteins. (B) Liver sections were immunolabelled with phalloidin to stain for F-actin (green) and with DAPI (blue) and visualized by confocal microscopy. Representative staining shows that actin microfilaments are arranged along the cell periphery in an organised pericanalicular pattern in control livers, but are grossly distorted and misshapen in α-catenin knockdown livers. Scale bar, 20 µm. (C-D) Representative images from electron microscopy performed on α-catenin knockdown and control livers. (C) Control BC consists of lumens filled with microvilli, as shown. Strikingly, the BC in α-catenin knockdown mice possess increased dilation in the lumens, with a severe paucity of microvilli. (D) A closer look at the tight junctions that seal the BC is shown in the magnified insets (bottom row). In control livers, tight junctions were marked by a fusion of two membranes into a characteristic darker single membrane. In contrast, there was an absence of fusion of the membranes on the sides of the BC in α-catenin knockdown livers, with tight junction regions exhibiting two separated plasma membranes. Scale bars, 0.2 µm.
We hypothesised that, if this disorganisation of actin filaments did indeed perturb the microvilli and contractibility of the canalicular membrane, the morphology of the bile canaliculi would be altered. Strikingly, electron microscopy showed highly dilated bile canalicular lumens in α-catenin knockdown livers, suggesting a diminished contractibility of the liver specimens. In addition, there was a severe reduction in the proportion of microvilli as compared with that observed in control livers (Fig. 4C). In control livers, tight junctions were marked by a distinct darker region comprising two apposed membrane bilayers. In contrast, in α-catenin knockdown livers, a space was observed between the plasma membranes on either side of the bile canaliculi, suggesting compromised integrity of the tight junctions (Fig. 4D). Hence, the loss of α-catenin resulted in the disorganisation of the actin filaments, leading to the loss of apical microvilli and dilated bile canaliculi with leaky tight junctions, which, in turn, resulted in a disruption to bile flow.
Finally, it was of interest to investigate the timeline of the defects brought about by the loss of α-catenin. Hence, α-catenin knockdown livers and blood samples were examined at different time points after partial hepatectomy: 1, 2, 4 and 6 days after surgery. The TBA and TBIL levels in the bloodstream of these mice were significantly elevated as early as day 1 after surgery, peaking at day 4 (Fig. 5A–B). Correlatively, the structure of the bile canaliculi was disorganised from day 1 after surgery, with observations of enlarged lumens that persisted until the day-6 time point (Fig. 5C). The liver-to-body weight ratio gradually increased in α-catenin knockdown mice from day 1, albeit with a significant up-regulation observed only from day 2 after surgery (Fig. 5D). In addition, nuclear immunolabeling of YAP was significantly higher in α-catenin knockdown livers from as early as day 1 and persisted through until day 6 (Fig. 5E). Additionally, CTGF expression was significantly up-regulated between days 1 and 2, which corresponds to the peak of hepatocyte proliferation (Fig. 5F). In all, these results suggest that the presence of α-catenin is crucial throughout the entire process of liver regeneration and that the loss of α-catenin causes chronic and persistent defects in the regenerative process.

α-catenin is required throughout the entire timeline of liver regeneration.
α-catenin siRNA-nanolipid particle (LNP)-injected mice were subjected to surgery and assessed at 1, 2, 4, 6 days after surgery for blood and liver analyses. (A) α-catenin siRNA-LNP-injected mice exhibited a significant increase in total bile acids from day 1 with a peak at day 4. (B) Total bilirubin levels (TBIL) showed a corresponding increase from day 1, again peaking at day 4. (C) ZO-1 staining (green) for bile canaliculi structure was disrupted in α-catenin siRNA-LNP injected mice as early as day 1 after surgery and persisted until day 6. (D) The total liver-to-body weight ratio was significantly increased only from day 2 (n = 3 for each time point except day 6; n = 2 for control group). All paired groups had significant p-values except day 6 (not significant). Scale bars, 20 µm. (E) Nuclei of cells from liver sections were immunolabelled with anti-YAP antibody (brown). The percentage of YAP-positive (+ve) nuclei was then calculated for α-catenin knockdown and control livers at each time point. Significant differences were measured at each time-point (p<0.05). (F) Gene expression of connective tissue growth factor (CTGF). CTGF was substantially up-regulated from day 2 onwards. Significant differences were measured at each time-point (p<0.05).
Discussion
Alpha-catenin has recently been uncovered to exert other functions aside from its role as a linker between cadherin and actin microfilaments13,25. In this study, we have unravelled a plethora of effects in liver regeneration attributed to the loss of α-catenin. Regenerated α-catenin knockdown livers were significantly larger than control regenerated livers, with the enlargement linked to an increase in hepatocyte proliferation. Similar increases in cell proliferation in response to the loss of α-catenin have been observed previously in other systems. Ablation of α-catenin in the mouse epidermis results in defects resembling the phenotype of squamous cell carcinoma, including hyperproliferation and altered cell polarity14. The hyperproliferation was found to be independent of cell adhesion and Wnt signalling, but connected to the Ras/MAPK pathway. In addition, a conditional knockout of α-catenin in the brain also causes hyperproliferation, which was attributed to the possible involvement of α-catenin in the Hedgehog pathway15. A previous study has also identified the involvement of β-catenin/Wnt signalling during the early stages of normal liver regeneration. Beta-catenin was shown to have an increased nuclear localisation as a result of its decreased degradation, which was thought to contribute to the increase in proliferation observed during regeneration26. In addition to these pathways, α-catenin has also been shown to negatively regulate Yap116, with the silencing of α-catenin resulting inthe relocalisation of YAP to the nucleus where it interacts with TEAD transcription factors to induce proliferation. More importantly, an overexpression of YAP drastically increases liver mass in mice27, which is similar to our findings here. One noteworthy point to bring up is that our ~6-fold increase in the percentage of Ki67-positive cells correlates with a 50% increase in liver mass, which is in line with their study that reports an almost 4-fold increase in percentage of Ki67-positive cells correlating with a 30% increase in liver mass, hence strengthening our data. Our results also point to α-catenin as a negative regulator of proliferation through distinct signalling pathways. It is highly plausible that the micro-environment dictates the degree of interaction between α-catenin and the different pathways. In our study, we found the increase in proliferation to be connected to the Hippo-YAP pathway. The loss of α-catenin in regenerated livers was accompanied by YAP activation, an increase in its nuclear translocation, as well as an increase in expression of CTGF, a direct YAP downstream target important for cell growth28. Notably, the increase in YAP nuclear translocation and CTGF expression occurred within the first two days of regeneration, corresponding to the peak proliferative period of hepatocytes. Hence, in accordance with previous studies, our results indicate that the loss of a-catenin results in an increase in the activation of YAP, which in turn, causes an increase in proliferation.
The liver-to-body mass ratio is consistently restored after the liver has regenerated3,5. Our observation of a greater liver-to-body mass ratio in α-catenin knockdown livers may imply that α-catenin plays a role in this master regulation. Hepatomegaly is present in many diseases, such as cirrhosis, fatty liver disease and in the obstruction of bile flow. Hence, the hepatomegaly observed in this case is possibly indicative of liver damage or malfunction. Previously, the activation or dysregulation of β-catenin has been shown to up-regulate the mTOR pathway in colon and ovarian tumours29,30. In our case, however, the increase in cell size was not attributed to an up-regulation of the mTOR pathway, as we did not observe any change in phosphorylated S6 kinase levels (data not shown). We also did not observe a significant change in β-catenin nuclear translocation in response to α-catenin knockdown, nor did we observe any significant increase in polyploidy, which could have contributed to cell hypertrophy (data not shown). Hence, the increase in cell size in our study is more likely due to dysregulation of the actin microfilaments, which could contribute to the change in cell size, perhaps through osmotic pressure changes31.
Normal bile formation and flow are important for proper homeostasis. The production of bile is crucial for the digestion of fat, the absorption of lipids and fat-soluble vitamins and the excretion of bilirubin, a by-product of recycled red blood cells. We found elevated levels of various plasma components, including TBIL, TBA, ALP and AMY, in our α-catenin knockdown mice, indicative of a defect in bile flow. Clinically, elevated TBA and TBIL levels are bona fide indicators of cholestasis, as is jaundice, which we observed in a subset of our α-catenin knockdown mice. Cholestasis occurs in various pathologies, such as primary liver cirrhosis, alcoholic liver disease, bile duct tumours and hepatitis and is characterised by a blockage of bile flow and therefore frequently associated with hepatomegaly, inflammation and decreased lipid levels. The phenotype observed in these α-catenin knockdown regenerated livers is characteristic of clinical cholestasis, with observations of disorganised bile canaliculi, hepatomegaly, elevated liver enzymes, bile salts and inflammation. Under conditions of cholestasis, a decrease in plasma HDL cholesterol is usually accompanied by an increase in plasma cholesterol; however, only HDL was affected in our α-catenin knockdown mice. This decrease in HDL likely derives from the increase in TBA, which, itself, activates the inhibitory FXR element on the promoter region of HDL32. FXR increases cholesterol ester transfer protein (CETP) activity, which transfers cholesterol from HDL to non-HDL particles33. Although there was no significant increase in total cholesterol, there was an increase in non-HDL particles, albeit an insignificant one.
The sinusoidal and bile canalicular networks of the regenerated α-catenin knockdown livers showed disorganised actin bundles at the pericanalicular domain, with tortuous and disorderly sinusoids. Also, the bile canaliculi exhibited dilated lumens with a loss of microvilli and compromised tight junction integrity. One hypothesis is that the loss of α-catenin is equivalent to the lack of a molecular switch to control the actin filaments13, resulting in actin cytoskeleton disruption and the ensuing perturbations to the bile canaliculi. Alpha-catenin is a central regulator of actin polymerisation in adherens junctions and acts as a mechanosensor and transducer in response to forces locally applied to this plasma membrane subdomain. In addition, zona occludens-1 (ZO-1), principally localised at the tight junction cytoplasmic domain, can also interact with α-catenin to locally control actin polymerisation34. Hence, it is not surprising that the loss of α-catenin resulted in the disorganisation of ZO-1. Any disruption in actin microfilament dynamics will have an impact on the stability of tight junctions, which, themselves, critically depend on the integrity of adherens junctions35. The lack of full-length α-catenin considerably affects the strength of the adhesion and prevents the expansion of the contact area in cell doublets36. Following these cytoskeletal changes, bile canaliculi in regenerating livers could develop abnormally dilated lumens with a loss of microvilli and leaky tight junctions. Notably, these cytoskeletal changes have been implicated in many human and animal cholestastic disorders37 and have been shown to cause a loss of apical microvilli and diminished contractibility of the bile canaliculi38. Moreover, an experimental induction of cholestasis in rats resulted in disruption of the tight junctions that seal the bile canaliculi39. This increased tight junction permeability might lead to an increase in paracellular permeability and a regurgitation of bile components back into the plasma. In line with this hypothesis, we observed a sharp increase in TBA and TBIL levels. In addition, cholestastic diseases, such as primary biliary cirrhosis and cholangitis, have been associated with inflammation. In our study, we measured an elevation in the total white blood cell count in the knockdown mice, indicative of an inflammatory response.
Interestingly, a recent study of the conditional deletion of β-catenin in the liver also resulted in bile canalicular abnormalities and intra-hepatic cholestasis40, albeit with a less severe phenotype than that described here. As mentioned earlier, there was no significant trend in β-catenin translocation associated with the loss of α-catenin. Hence, it is intriguing to consider that the effect of α-catenin loss in our system is independent of its role in cell adhesion. The role of E-cadherin, which is expressed in the liver, is unclear based on seemingly conflicting reports. In monolayer cultures of hepatocytes, E-cadherin was shown to be important for the maintenance of extended networks of bile canaliculi41. In β-catenin conditional liver-specific knockout mice, no change in tight junction morphology or bile canaliculi appearance was observed42. In all, the lack of a similar severe phenotype in these studies strengthens the case that the well-known role of α-catenin in cell adhesion is not the role critical for the phenotype observed.
The silencing of α-catenin expression in normal homeostatic liver incurred a more modest phenotype, with slight alterations in BC morphology and liver enzymes, but no change in liver mass. Hence, this shows that α-catenin is especially important during a time period of rapid proliferation and formation of new structures during the regenerative period, rather than during a senescent state of the liver. Indeed, the presence of α-catenin seems to be crucial and necessary for normal liver regeneration and its loss appears to affect the process acutely and persistently. We observed an increase in cell proliferation and bile canalicular defects in all α-catenin knockdown livers harvested at various time points. Alpha-catenin knockdown induced a striking phenotype of a grossly enlarged liver attributed partially to hepatomegaly and partially to increased proliferation and severe defects in bile canalicular formation, changes that mimic the clinical condition of cholestasis. Compared with its known interacting partners, which exhibit relatively modest phenotypes when affected, α-catenin appears to be important in numerous signalling pathways connected with normal liver function, liver regeneration and cholestasis. By understanding the role of α-catenin in these processes, we can shed light on the associated pathologies and develop prospective corresponding treatments.
Methods
Injection of siRNA-nanolipids
Animal protocols were approved by the Institutional Animal Care and Use Committee at A*STAR, Singapore and conformed to National Institutes of Health guidelines and public law. All experiments were performed in accordance with these approved animal protocols. siRNA targeting transcripts of alpha-catenin were synthesized by Axolabs (Kulmbach, Germany). 2'OMe modified nucleic bases and dTsdT 3' overhangs (‘s’ indicates phosphorothioate linkage) were introduced to increase siRNA in vivo stability and inhibit immune stimulation. siRNAs were formulated into lipidoid nanoparticles (LNP) targeting preferentially the liver, with a particle mean diameter of ~60–80 nm43,44,45,46. In order to ensure significant knockdown of each gene at both mRNA and protein levels, 9- to 11-week-old Balb/cByJ female mice were injected via the tail vein with LNP at a dose of 0.5 mg/kg body weight every 5 days for 20 days, with an additional injection performed at day 27.
Partial hepatectomy
Partial hepatectomy was performed on the animals on day 25, as previously described4. Dissected lobes were fixed in 10% neutral-buffered formalin or fresh-frozen on dry ice. Fresh tissue was also frozen-down for protein and RNA analyses. Seven days after surgery, the mice were sacrificed and the livers were harvested for analysis.
Cell culture
AML12 cells were purchased from ATCC (Manassas, VA, USA) and cultured according to ATCC recommended conditions. Transfection was performed using Lipofectamine 2000 (Life Technologies) with ON-TARGETplus mouse α-catenin siRNA (Thermo Fisher Scientific),
Hepatocyte isolation, seeding and culturing
Hepatocytes were isolated from male Wistar rats by a two-step in-situ collagenase perfusion method. Animals were handled according to the IACUC protocol approved by the IACUC committee of the National University of Singapore. With a yield of >108 cells/rat, viability of the hepatocytes was tested to be >90% by Trypan Blue exclusion assay. Freshly isolated rat hepatocytes (0.2 million) were seeded onto 24-well plate and cultured in 0.5 ml of William’s E culture medium supplemented with 2 mM L-Glutamine, 1 mg/ml BSA, 0.3 μg/ml of insulin, 100 nM dexamethasone, 50 μg/ml linoleic acid, 100 units/ml penicillin and 100 mg/ml streptomycin, all of which were purchased from Aldrich-Sigma. Cells were incubated with 5% CO2 at 37°C and 95% humidity. After 3-hour incubation, the unattached cells were removed. Culture medium was changed on a daily basis. 0.25 mL 0.5 mg/ml neutral collagen was added into each well 24 hours after cell seeding and incubated at 37°C for 3 hours. Fresh medium was added after gelation of collagen.
Immunohistochemistry
Tissues fixed in formalin were either embedded in Tissue-Tek (Sakura Finetek Europe B.V, The Netherlands) or embedded in paraffin and sectioned accordingly. The fresh-frozen sections were fixed in ethanol at −20°C for 30 min. The paraffin sections were de-paraffinised, rehydrated and subjected to antigen retrieval using a sodium citrate buffer. They were blocked using 2.5% bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA) and 0.3% Triton X-100 (VWR, Poole, England) for 2 h, after which the sections were incubated with primary antibody solution overnight at 4°C, followed by incubation with the appropriate secondary antibody for 1.5 h. The slides were then stained with DAPI and mounted with Vectashield (Vectorlabs, Burlingame, CA, USA). The antibodies used were rabbit anti-N-cadherin (ab18203; Abcam, Cambridge, MA, USA), rabbit anti-collagen IV (ab6586; Abcam), rabbit anti-YAP1 (sc-15407; Santa Cruz, Dallas, TX, USA), rat ZO-1 (DSHB, Iowa City, IA, USA), rabbit anti-Ki67 (ab15580; Abcam) and phalloidin-AF488 (A12379; Life Technologies, Carlsbad, CA, USA). Primary antibodies were detected with AF546-conjugated and AF488-conjugated secondary antibodies (Life Technologies). Imaging was performed using a fluorescence microscope (CTR6000; Leica, Wetzlar, Germany) or a confocal microscope (LSM700; Zeiss, Oberkochen, Germany).
Immunofluorescence and image acquisition
Hepatocytes or AML12 cells were fixed in 4% paraformaldehyde (PFA) for 30 min and permeabilized for 30 min in TBST (0.2% Triton-X in TBS). The permeabilized cells were blocked with 1% BSA in TBST for 4 h and incubated with phalloidin-Alexa 488 (Life Technologies) for 1 h in the dark at room temperature. After staining with DAPI (Sigma-Aldrich), cells were mounted in mounting medium (DAKO, Carpinteria, CA, USA). A 3D stack of confocal microscopy images were acquired with 100× NA1.4 oil lens on a Nikon A1R Confocal Microscope.
Blood chemical analysis
Blood was collected via cardiac puncture using a 21-G needle under anaesthesia. A haematology panel (HEMAVET 950FS, Thermo Fisher Scientific, Waltham, MA, USA) and a blood chemistry panel of serum markers and enzymes (VETSCAN VS2, Abaxis, Union City, CA, USA) were analysed using automated methods in the Histopathology Department of the Institute of Molecular Cell Biology, A*STAR. Total bile acid levels in serum were analysed using a commercially available kit from Diazyme (Poway, CA, USA) and each sample was tested in triplicate. Phospholipids, triglycerides and total cholesterol levels in plasma were determined using commercially available kits from Biovision (Mountain View, CA, USA), Wako Chemicals (Richmond, VA, USA) and Stanbio (Boerne, TX, USA), respectively,
Ki67 and YAP quantification
Each sample was double-labelled with an anti-Ki67 antibody and DAPI and a total of 20 images were taken at 40× magnification. Similarly, each sample was labelled with anti-YAP and images were taken at 20× magnification. The numbers of Ki67-positive or YAP-positive nuclei in each field were quantified using ImageJ software (NIH, Bethesda, MD, USA). The percentage of Ki67-positive or YAP-positive nuclei was then calculated by dividing the total number of Ki67-positive or YAP-positive nuclei by the total number of nuclei.
Statistical tests and quantification
All p-values were calculated using an unpaired t-test with a two-tailed distribution and assuming unequal variance. For the cell size quantification and the liver simulation model, cells were labelled with a membrane marker to visualize the outline of the cells. A rotational kernel was used to enhance the membrane signal and the images were converted to binary images. A distance transform formula was applied and the local maximum was set as the centre of the cell. Based on this cell centre, Evolving Generalized Voronoi Diagram47,48 method was applied to segment the cells. Based on the segmentation results, the probability distribution function was created for each condition. In order to determine if the increase in liver size was due to an increase in cell size, a numerical simulation was conducted. A simulation model was created for both control and siRNA-injected cells, by taking into consideration 1 million cells for each group and generating the probability density function for each of them.
RNA extraction and first strand cDNA synthesis
RNA was extracted from frozen mouse livers and monolayer cells using the Qiagen RNeasy mini kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's protocol. First-strand cDNA synthesis was carried out using a Superscript III reverse transcriptase kit (Life Technologies).
Real-time PCR
Real-time PCR with SYBR green detection or Taqman gene expression assays was used to confirm knockdown and evaluate the expression levels of various genes from both cell cultures and tissue samples, performed with the following primers: E-cadherin F: CAG CCT TCT TTT CGG AAG ACT, R: GGT AGA CAG CTC CCT ATG ACT G. α-catenin F: TCT CTA CTG CCA CCA GCT CAA C, R: AAG CCA TCC CCT GTG ACT TCT. MRP2 F: GTG TGG ATT CCC TTG GGC TTT, R: CAC AAC GAA CAC CTG CTT GG MRP3 F: CTG GGT CCC CTG CAT CTA C, R: GCC GTC TTG AGC CTG GAT AAC MDR1: Quantitect primers QT01051988, MDR2: Quantitect primers QT00164010 (Qiagen, Venlo, The Netherlands). CTGF: Mm01192932_g1, ACTIN: Mm00487499 (Life Technologies). Appropriate non-template controls were included in each run.
Electron microscopy
Liver tissue samples were fixed in a 0.1 M phosphate buffer, pH 7.4, containing 4% PFA, 2.5% GA and 0.2% picric acid and then post-fixed in 0.1 M sodium cacodylate trihydrate, pH 7.6, comprising 1% osmium tetroxide and 1.5% potassium ferricyanide. Samples were then dehydrated, stained with 2% uranyl acetate in 75% ethanol, infiltrated with acetone and embedded in resin. The blocks were then trimmed to remove excess resin. An ultramicrotome was used to create 0.5-µm semi-thin sections, which were stained with methylene blue with Borax. After identifying the area of interest, each sample was then trimmed down to an area less than 3 mm. Ultrathin sections of 90-nm were cut and collected with 150 mesh copper grids. Grids were then stained with heavy metal stains, uranyl acetate (10 min) and lead citrate (8 min), for contrast. Viewing and image acquisition were performed on the Jeol 1220 TEM (Tokyo, Japan).
Western blot
Liver tissues were homogenized on ice for 5 sec and then lysed in RIPA buffer containing 2 mM/ml Na3VO4 and 1x protease inhibitor cocktail (Sigma- Aldrich) for 20 min at 4°C. Whole tissue lysates were separated using SDS-PAGE and transferred onto a PVDF membranes. Primary antibodies used were Alpha-catenin (610194; BD Biosciences, San Jose, CA, USA), GAPDH (ab9485; Abcam), Yap (4912; Cell Signaling, Danvers, MA, USA) and phosphor-Yap-Ser127 (4911; Cell Signaling).
References
Foundation, A. L. Liver Wellness. Increasing Public Awareness of Liver Health, <http://www.liverfoundation.org/downloads/alf_download_29.pdf> (2009). Accessed 7th July 2014.
Liver, E. A. f. t. S. o. Facts and Figures. The Liver: Vital for Life, <http://old.easl.eu/eu_facts.asp> (2009). Accessed 7th July 2014.
Huang, J. & Rudnick, D. A. Elucidating the metabolic regulation of liver regeneration. Am. J. Pathol. 184, 309–321, 10.1016/j.ajpath.2013.04.034 (2014).
Mitchell, C. & Willenbring, H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 3, 1167–1170, 10.1038/nprot.2008.80 (2008).
Michalopoulos, G. K. Liver regeneration. J. Cell. Physiol. 213, 286–300, 10.1002/jcp.21172 (2007).
Yonemura, S., Wada, Y., Watanabe, T., Nagafuchi, A. & Shibata, M. Alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell. Biol. 12, 533–542, 10.1038/ncb2055 (2010).
Hansen, S. D. et al. alphaE-catenin actin-binding domain alters actin filament conformation and regulates binding of nucleation and disassembly factors. Mol. Biol. Cell 24, 3710–3720, 10.1091/mbc.E13-07-0388 (2013).
Borghi, N. et al. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. U.S.A 109, 12568–12573, 10.1073/pnas.1204390109 (2012).
Hong, S., Troyanovsky, R. B. & Troyanovsky, S. M. Binding to F-actin guides cadherin cluster assembly, stability and movement. J. Cell Biol. 201, 131–143, 10.1083/jcb.201211054 (2013).
Abe, K. & Takeichi, M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl. Acad. Sci. U.S.A 105, 13–19, 10.1073/pnas.0710504105 (2008).
Thomas, W. A. et al. Alpha-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 288, 4957–4969, 10.1074/jbc.M112.403774 (2013).
Pokutta, S., Drees, F., Takai, Y., Nelson, W. J. & Weis, W. I. Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J. Biol. Chem. 277, 18868–18874, 10.1074/jbc.M201463200 (2002).
Drees, F., Pokutta, S., Yamada, S., Nelson, W. J. & Weis, W. I. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 123, 903–915, 10.1016/j.cell.2005.09.021 (2005).
Vasioukhin, V., Bauer, C., Degenstein, L., Wise, B. & Fuchs, E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell 104, 605–617 (2001).
Lien, W. H., Klezovitch, O., Fernandez, T. E., Delrow, J. & Vasioukhin, V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 311, 1609–1612, 10.1126/science.1121449 (2006).
Schlegelmilch, K. et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144, 782–795, 10.1016/j.cell.2011.02.031 (2011).
Shtutman, M. et al. Signaling function of alpha-catenin in microtubule regulation. Cell Cycle 7, 2377–2383 (2008).
Malato, Y. et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest. 121, 4850–4860, 10.1172/JCI59261 (2011).
Akinc, A. et al. Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol. Ther. 17, 872–879, 10.1038/mt.2009.36 (2009).
Bogorad, R. L. et al. Nanoparticle-formulated siRNA targeting integrins inhibits hepatocellular carcinoma progression in mice. Nat Commun 5, 3869, 10.1038/ncomms4869 (2014).
Speicher, T. et al. Knockdown and knockout of beta1-integrin in hepatocytes impairs liver regeneration through inhibition of growth factor signalling. Nat Commun. 5, 3862, 10.1038/ncomms4862 (2014).
Wolfrum, C. et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 25, 1149–1157, 10.1038/nbt1339 (2007).
Zollner, G. & Trauner, M. Molecular mechanisms of cholestasis. Wien. Med. Wochenschr. 156, 380–385, 10.1007/s10354-006-0312-7 (2006).
Trauner, M., Meier, P. J. & Boyer, J. L. Molecular pathogenesis of cholestasis. N. Engl. J. Med. 339, 1217–1227, 10.1056/NEJM199810223391707 (1998).
Maiden, S. L. & Hardin, J. The secret life of alpha-catenin: moonlighting in morphogenesis. J. Cell Biol. 195, 543–552, 10.1083/jcb.201103106 (2011).
Monga, S. P., Pediaditakis, P., Mule, K., Stolz, D. B. & Michalopoulos, G. K. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology 33, 1098–1109, 10.1053/jhep.2001.23786 (2001).
Dong, J. et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133, 10.1016/j.cell.2007.07.019 (2007).
Zhao, B. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971, 10.1101/gad.1664408 (2008).
Tanwar, P. S. et al. Mammalian target of rapamycin is a therapeutic target for murine ovarian endometrioid adenocarcinomas with dysregulated Wnt/beta-catenin and PTEN. PLOS One 6, e20715, 10.1371/journal.pone.0020715 (2011).
Fujishita, T., Aoki, K., Lane, H. A., Aoki, M. & Taketo, M. M. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proc. Natl. Acad. Sci. U.S.A 105, 13544–13549, 10.1073/pnas.0800041105 (2008).
Tinevez, J. Y. et al. Role of cortical tension in bleb growth. Proc Natl Acad Sci U S A 106, 18581–18586, 10.1073/pnas.0903353106 (2009).
Claudel, T. et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J. Clin. Invest. 109, 961–971, 10.1172/JCI14505 (2002).
Gautier, T. et al. Farnesoid X receptor activation increases cholesteryl ester transfer protein expression in humans and transgenic mice. J. Lipid Res. 54, 2195–2205, 10.1194/jlr.M038141 (2013).
Yonemura, S. Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell. Biol. 23, 515–522, 10.1016/j.ceb.2011.07.001 (2011).
Huang, R. Y., Guilford, P. & Thiery, J. P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 125, 4417–4422, 10.1242/jcs.099697 (2012).
Engl, W., Arasi, B., Yap, L. L., Thiery, J. P. & Viasnoff, V. Actin dynamics modulate mechanosensitive immobilization of E-cadherin at adherens junctions. Nat Cell Biol 16, 587–594, 10.1038/ncb2973 (2014).
Phillips, M. J., Poucell, S. & Oda, M. Mechanisms of cholestasis. Lab. Invest. 54, 593–608 (1986).
Anderson, J. M. Leaky junctions and cholestasis: a tight correlation. Gastroenterology 110, 1662–1665 (1996).
Rahner, C., Stieger, B. & Landmann, L. Structure-function correlation of tight junctional impairment after intrahepatic and extrahepatic cholestasis in rat liver. Gastroenterology 110, 1564–1578 (1996).
Yeh, T. H. et al. Liver-specific beta-catenin knockout mice have bile canalicular abnormalities, bile secretory defect and intrahepatic cholestasis. Hepatology 52, 1410–1419, 10.1002/hep.23801 (2010).
Terry, T. L. & Gallin, W. J. Effects of fetal calf serum and disruption of cadherin function on the formation of bile canaliculi between hepatocytes. Exp. Cell Res. 214, 642–653, 10.1006/excr.1994.1302 (1994).
Battle, M. A. et al. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc. Natl. Acad. Sci. U.S.A 103, 8419–8424, 10.1073/pnas.0600246103 (2006).
Love, K. T. et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. U.S.A 107, 1864–1869, 10.1073/pnas.0910603106 (2010).
Akinc, A. et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 18, 1357–1364, 10.1038/mt.2010.85 (2010).
Frank-Kamenetsky, M. et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A 105, 11915–11920, 10.1073/pnas.0805434105 (2008).
Zeigerer, A. et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 485, 465–470, 10.1038/nature11133 (2012).
Yu, W., Lee, H. K., Hariharan, S., Bu, W. & Ahmed, S. Evolving generalized Voronoi diagrams for accurate cellular image segmentation. Cytometry A 77, 379–386, 10.1002/cyto.a.20876 (2010).
Yu, W., Lee, H. K., Hariharan, S., Bu, W. & Ahmed, S. Quantitative neurite outgrowth measurement based on image segmentation with topological dependence. Cytometry A 75, 289–297, 10.1002/cyto.a.20664 (2009).
Acknowledgements
We thank the Histopathology Department of our Institute for blood analysis and histology work. This work was supported by the Agency of Science, Technology and Research (A*STAR).
Author information
Authors and Affiliations
Contributions
K.J.H. designed the experiments, carried them out and wrote the paper. N.T. assisted in the animal experiments and performed the western blots. J.W.E.O. assisted in the experiments. Q.L. and V.V. performed the experiments involving primary rat hepatocytes. L.L.Y. provided preliminary data leading to the study. H.Y., R.B., J.E.D., D.G.A. and V.K. formulated and provided the siRNA-LNPs. W.Y. executed the statistical analysis of cell size and the liver simulation model. Y.G.C. and B.H.B. performed the electron microscopy. R.S. provided lipid analysis of the serum samples. J.P.T. provided advice and directions to the study and edited the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary figures
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Herr, K., Tsang, Yh., Ong, J. et al. Loss of α-catenin elicits a cholestatic response and impairs liver regeneration. Sci Rep 4, 6835 (2014). https://doi.org/10.1038/srep06835
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06835
This article is cited by
-
Hippo signalling in the liver: role in development, regeneration and disease
Nature Reviews Gastroenterology & Hepatology (2022)
-
Mechanoregulation and pathology of YAP/TAZ via Hippo and non‐Hippo mechanisms
Clinical and Translational Medicine (2018)
-
The Hippo pathway in intestinal regeneration and disease
Nature Reviews Gastroenterology & Hepatology (2016)
-
Curcumin protects ANIT-induced cholestasis through signaling pathway of FXR-regulated bile acid and inflammation
Scientific Reports (2016)
-
αE-catenin inhibits YAP/TAZ activity to regulate signalling centre formation during tooth development
Nature Communications (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
