
Abstract
It has been recently demonstrated that the internal jugular vein may exhibit abnormalities classified as truncular venous malformations (TVMs). The investigation of possible morphological and biochemical anomalies at jugular tissue level could help to better understand the link between brain venous drainage and neurodegenerative disorders, recently found associated with jugular TVMs. To this end we performed sequential X-ray Fluorescence (XRF) analyses on jugular tissue samples from two TVM patients and two control subjects, using complementary energies at three different synchrotrons. This investigation, coupled with conventional histological analyses, revealed anomalous micro-formations in the pathological tissues and allowed the determination of their elemental composition. Rapid XRF analyses on large tissue areas at 12.74 keV showed an increased Ca presence in the pathological samples, mainly localized in tunica adventitia microvessels. Investigations at lower energy demonstrated that the high Ca level corresponded to micro-calcifications, also containing P and Mg. We suggest that advanced synchrotron XRF micro-spectroscopy is an important analytical tool in revealing biochemical changes, which cannot be accessed by conventional investigations. Further research on a larger number of samples is needed to understand the pathogenic significance of Ca micro-depositions detected on the intramural vessels of vein walls affected by TVMs.
Similar content being viewed by others

Introduction
Tuncular venous malformations (TVMs) are the result of the developmental problems of vascular trunk formation during the fetal phase, TVMs are divided into obstruction and dilation (aneurysms)1. A relevant cause of obstructive TVMs are intraluminal obstacles such as septa, webs, membranes, fixed and rudimental valves, or wall stenosis (segmentary hypoplasia). TVMs may have different hemodynamic impacts on their relevant drained apparatus/organ. Independently from the area where they occur, the impact is chronic and progressive in the clinical course, depending upon their location, extent/severity and natural compensation through collaterals1.
For instance, membranous obstruction of the inferior vena cava in primary Budd-Chiari syndrome is a classic example of congenital TVM determining a focal segmental occlusion of the suprahepatic inferior vena cava. This disorder can lead to profound portal hypertension due to hepatic venous outlet obstruction with severe consequences consisting in chronic hepatic failure and liver sclerosis1,2,3.
Moreover, in the last years it has been described TVMs characterized by intraluminal obstacles at the level of the internal jugular vein (IJV), or sometimes, by segmentary hypoplasia.
Collectively, especially if involved also the azygous vein, these IJV abnormalities determine a restricted venous outflow from the brain and from the spine. This condition is named chronic cerebrospinal venous insufficiency (CCSVI). The reported prevalence of CCSVI is highly heterogeneous in literature. However, a meta-analyses reveals that CCSVI was found mainly in multiple sclerosis (MS) patients, but also possibly associated to other neurodegenerative diseases and even to healthy controls4,5,6 CCSVI was found to be associated to other neurodegenerative disorders, out of MS, such as Alzheimer7, Meniere8,9 and Parkinson syndrome10. However, little is known about the pathology characterizing the jugular venous wall in this condition.
Independently from the association with MS and/or other neurodegenerative disorders, CCSVI is linked to reduced perfusion11,12,13. Moreover, another significant change in brain pathophysiology associated with CCSVI is the reduction of cerebro-spinal fluid flow, as a consequence of impaired reabsorption14,15.
To improve knowledge of the underlying biochemical mechanisms involved in such a novel vascular disease, with potential implications in the process of neurodegeneration, the aim of our research was to investigate the chemical changes occurring in the jugular tissue of CCSVI patients. In particular, as a possible additional approach to conventional techniques, we experimented with the potential of synchrotron radiation based X-ray Fluorescence (XRF) microscopy at different incident energies to reveal characteristic chemical features of pathological tissues. X-ray fluorescence analysis is a multi-elemental, highly sensitive technique based on the detection of X-rays emitted from samples' atoms excited with X-ray photons. This technique can provide semi-quantitative or quantitative information since the fluorescence intensity is related to the concentration of the element within the sample. Over the past decade, the investigation of biological samples with XRF has been boosted by the development of high-flux and highly focused X-ray beam at different synchrotron facilities16,17,18.
Based on these considerations, we have carried out a pilot study to explore the ability of three XRF set-ups at different spatial resolutions and at different energies to reveal elemental changes into CCSVI jugular vein specimens.
Results
Histological analyses of the jugular samples
Samples obtained from jugular specimens of MS patients and control subjects were morphologically and histologically analyzed to evaluate the presence of alterations of the vein wall. Careful histological examination of several sections of MS jugular veins wall after hematoxylin and eosin (HH) staining demonstrated the presence of small basophilic bodies suggestive of micro-calcifications within the connective tissue of the tunica adventitia. These structures were often seen in relationship with the lumen of the vasa vasorum. Some sections showed shredding of the tissue due to the presence of hard calcified material causing resistance to the microtome during cutting. No analogous structures were found in the control samples.
As shown in Figure 1, histological analysis of the MS samples revealed the presence of micro-calcifications appearing as small amorphous bodies stained blue-gray with hematoxylin, placed between the collagen bundles of the tunica adventitia of the vein wall (panel b). These structures were frequently clearly located inside the lumen of the vasa vasorum microvessels (panels c and d). Panel (a) shows a tear in a tissue section along with residual fragments of basophilic material, due to a sectioning artifact. To further explore the presence and nature of the micro-depositions in MS jugular samples, von Kossa stain and Alizarin red test were performed. However these techniques produced negative results in all the investigated tissues.

Optical microscopy images of anomalies in MS jugular vein tissues.
Images a and b show suggested micro-calcifications (arrows). A scratch in the tissue is evident in a, while b, c and d show the singular appearance of same microvessel. The arrow in c indicates the presence of basophilic-calcified material inside a capillary. The same is revealed in panel d. All images are at 40 × magnification.
In morphological analysis, performed by SEM (Supplementary Figure S1), the control vein showed a virtually intact endothelial layer, with regular arrangement of the cells. This appearance changed significantly in pathological specimens, which displayed areas of partially detached endothelial cells and the loss of the integrity of the luminal monolayer as evidenced by craters or cavities (Supplementary Figure S1).
Synchrotron XRF analysis of the jugular samples
MS and control samples were analysed by XRF performed at different spatial resolution and at complementary energies on several sequential sections of paraffin-embedded tissues. Results were then compared to histological evaluations, in order to assess the presence of possible chemical changes corresponding to morphological alterations of the vein wall, as suggested by HH staining.
Unstained tissue sections (from 3 to 5 sections for each tissue) were initially analyzed by the XRF set-up of the XFM beamline of the Australian Synchrotron that is suitable for rapid analysis of large tissue areas.
Supplementary Figure S2 clearly shows the potential of the Maia detector, where areas of around 4 mm2 were analysed in approximately 3–5 hours. This amazingly fast screening of the tissues proved to be very effective in revealing the presence and the distribution of Zn, Fe and Ca, the latter being the lightest element detectable with the current version of the Maia detector. After deconvolving the point by point spectra of each map, the Geopixe software was used to calculate the concentration of the elements of interest (Ca, Fe and Zn) (see Supporting Information). It was observed that Ca concentration tended to be higher in all the sections of MS tissues compared to the control samples. To better understand this initial observation, the large maps were divided in smaller regions that allow association of elemental distribution and particularly the increased Ca concentration with specific tissue structures.
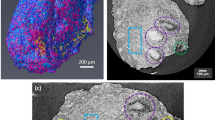
As shown in Figure 2, the XRF analyses at 12.74 keV on the MS2 sample sections reveal that the high Ca concentration is mainly associated with microvessels inside the tissue. The elemental maps are acquired on consecutive tissue slices of MS2 sample, on the regions indicated in the corresponding stained sample (panels a and b).

XRF elemental maps at 12.74 keV in MS2 tissue jugular sections.
Three consecutive tissue sections of MS2 sample are used: two unstained for XRF analyses and one HH stained for tissue structure recognition. a) and b): light microscopy images, the boxes indicate the selected regions for XRF analyses in the unstained sections. The corresponding elemental maps of Ca, Fe and Zn of regions 1, 2 and 3 acquired at the XFM beamline at 12.74 keV with 2 μm spatial resolution on the corresponding unstained tissue slices are shown in rows 1, 2 and 3 respectively. Red arrow in Ca map (1) indicates a calcification further analyzed at 4.12 keV. Arrows in Zn map indicate potential contaminants and tissue debris. The concentrations reported on the scale bars are in ppm. Region 1: 250 μm × 170 μm; Region 2: 600 μm × 400 μm; Region 3: 700 μm × 600 μm.
In all the areas there is a clear co-localisation of Fe and Zn that mainly corresponds to the presence of red blood cells and capillaries19. While Zn concentration ranges from 10 to 200 ppm in the tissue, broader ranges can be found for Fe and Ca. The maximal concentration of Fe is related to the presence of blood, while Ca hotspots are located in small areas suggesting microcalcifications. In region 1, Ca appears to be almost exclusively present on the microvessel walls in spots of variable size from a few to tens of microns. Almost the same appears in regions 2 and 3, even though Ca hotspots are present also in the surrounding tissue. Some Ca and Zn co-localizations in region 3 seem to reveal contaminants or tissue debris possibly due to the slicing that is visible in the stained tissue (panel b).
In order to better investigate Ca distribution especially in the vasa venorum, high Ca regions were analyzed at lower energy and higher spatial resolution at the ID21 (7.2 keV) and TwinMic (1.5 keV) beamlines. The set up of the ID21 microscope allows detection of the distribution of P and S together with Fe and Ca, while the TwinMic station highlights the location of light elements in addition to Fe.
Figure 3 depicts some selected microvessels (as indicated in the visible light image VL) previously analysed at 12.74 keV and shown in Figure 2 (region 2). With the ID21 and TwinMic set-ups absolute quantification was not obtained and the scale bar graduations simply show relative XRF intensities inside the single maps. In Figure 3, in both areas 1 and 2, the Fe distribution reflects the presence of blood, while S, still extremely high in blood, better delineates tissue structures, specially allowing demarcation of the endothelial border of the small vessel. Generally Ca and P could be related to some cell structures, particularly when co-localizing with S. However the Ca concentration is highest in some segments of the endothelium, which are also high in P but poor in S. These structures are compatible with calcifications. Figure 3, region 3, shows low energy XRF (LE-XRF) analyses acquired at 1.5 keV where the light elements C, O, Na and Mg can be correlated to the absorption and phase contrast images of a sub-area of region 2. The absorption and phase contrast images are used to recognize the morphology of the tissue structures. The first imaging modality is sensitive to tissue density and even more to the presence of iron, which is indeed the heaviest element (dark in the absorption image); the latter instead correlates to tissue topography, being more sensitive to discontinuities in the specimen such as borders, edges and protuberances. The most interesting elements in these images are Na and Mg, the first revealing the endothelial morphology, the second appearing substantially correlated to the Ca distribution in region 2. Interestingly, in this case the regions with the highest Mg signal are lower in C and O, thus this further suggests the presence of phosphate calcifications also containing Mg. This is particularly evident in the suspected calcification of around 10 μm size also shown in the Ca map of region 2 by arrow.

XRF analyses of microvessels in a MS2 jugular tissue section.
Row 1: elemental maps of Ca, P, S and Fe acquired on regions 1 (100 μm × 54.5 μm) at 7.2 keV (ID21 beamline) with 0.5 μm spatial resolution and 300 ms/pixel acquisition time; Row 2: elemental maps of Ca, P, S and Fe acquired on region 2 (100 μm × 76 μm) at 7.2 keV (ID21 beamline) with 0.5 μm spatial resolution and 300 ms/pixel acquisition time; Row 3: Absorption (Abs) and phase contrast images (PhC) with the corresponding elemental maps of C, O, Mg and Na collected on region 3 (80 μm × 80 μm) at 1.5 keV (Twinmic beamline) with 0.5 μm spatial resolution and 10 s/pixel acquisition time. The analysed regions are indicated in the corresponding visible light image (VL) of the MS2 tissue section. The red arrow indicates a region analysed at 4.12 keV too.
The results of similar investigations on sample MS1 are shown in Figure 4. Regions 1 and 2 correspond to microvessels showing Ca deposition on the endothelial walls, while region 3 suggests a calcification close to the luminal side of the jugular specimen. As in Figure 2, at 12.74 keV energy (regions 1 and 2) the co-localization of Fe and Zn is useful for indentifying the presence of microvessel in the unstained tissues. The Ca concentration signal reaches values up to 2000 ppm on and around the microvessel walls. The analyses at 7.2 keV for the sub-region 2b confirm, as for patient MS2, the abundant co-localization of Ca and P on the endothelial cell layer. In this particular region it is interesting that a diffuse low level Fe signal is found around the microvessel. In region 3 the sulfur map (panel S) mainly delineates the tissue morphology since it is extremely high in the extracellular matrix. In addition, it reveals the borders of the endothelial layer, while Fe, P and Ca have quite complementary distributions. Close to the endothelial side, Fe, P and Ca are mainly co-localized in a triangular structure (around 40 μm wide) where S is lower in signal. The morphology, the composition and signal intensities are compatible with a calcification, in which, for this case, there is also an unusually high abundance of iron. A similar iron concentration associated with increased Ca and P has been found also in other micro-calcifications in both MS tissues in a few rare cases. The presence of Fe in micro-calcifications in other tissues is however reported by other authors20.

XRF analyses in sample MS1.
VL: visible image of a stained slice of MS1 sample: boxes represent the corresponding sub-areas resolved under XRF analyses. 1 and 2: Ca, Fe and Zn XRF map (150 μm × 160 μm) acquired at the XFM beamline. The concentration is in ppm. Figures 2b: elemental maps of S, P, Fe and Ca in a sub-area (30 μm × 40 μm) of Panel 2 investigated at 7.2 keV (ID21 beamline). Area 3: elemental maps of S, P, Fe and Ca (160 μm × 60 μm) investigated at 7.2 keV (ID21 beamline).
In Figure 5 we report the XRF analyses of two control tissues at three different energies. Both regions contain normal vasa vasorum. All the analyses are performed with the same set-ups and acquisition conditions as for MS patients. In order to provide the distribution maps of the same elements as for the MS samples (Figure 4), phase contrast images and C, O and Na maps for the control samples shown in Figure 5 are depicted in Supplementary Figure S3 of supporting information. From the analyses at 12.74 keV in samples V1and V2, Ca concentration in the endothelial tissues reaches values up to 200 ppm, more than an order of magnitude less than in the MS tissues (data not shown). Zinc and Fe maps are useful to identify microvessels (panels a). Sub-areas of the region in panels a were analyzed at 7.2 keV (b rows) and reveal a greatly reduced Ca presence compared to that of Figures 3 and 4. The most striking result is the clear difference in the Ca/P XRF intensity ratio (counts) in the co-localization areas. Similarly, from the analyses at 1.5 keV, Mg intensity (panels c) is lower than in MS tissues. By comparing the Na and Mg maps for MS (Figure 3) and control samples (Figure 5 and Supplementary S3) it can be noticed that Mg concentration is higher in the MS than in control tissues, suggesting that the calcification could contain magnesium. An interesting result is obtained by comparing Mg to Na distribution. When the Mg/Na counts ratio (ratio between peak counts) is calculated on calcifications of MS samples (more than 20 points tested) it ranges from 1.7 to 1.9, while in calcification-free regions and in healthy tissue it varies from 0.15 to 1.3 (and is even below 1 at vena venorum level in the control samples).

XRF analysed of microvessels in control tissues.
Panels a: Elemental maps of Zn and Fe acquired at 12.74 keV and 2 μm spatial resolution (a-V1 area: 600 μm × 450 μm, 15 ms/pixel; a-V2 area: 300 μm × 225 μm, 15 ms/pixel). Panels b: Elemental maps of S, Ca, Fe and P acquired at 7.2 keV and 0.5 μm spatial resolution (B-V1 area: 80 μm × 70.5 μm; b-V2 area: 100 μm × 100 μm both 300 ms/pixel). Panels c: Absorption (Abs) image and corresponding Mg elemental map investigated at 1.5 keV and 0.5 μm spatial resolution (both areas 80 μm × 80 μm, 10 s/pixel).
The Ca/P ratio on calcifications
To better highlight the differences in terms of Ca content between the vasa vasorum of MS samples and those of controls, we conducted analyses at 4.12 keV using the ID21 set-up. At this energy the efficiency of XRF emission for Ca, P and S is higher thus providing better detection limits for all of them. Figure 6 shows some structures selected for the high Ca concentration accordingly to the previous analyses at higher energies: a corresponds to a sub-region of 30 μm × 30 μm from Figure 2, region (1) (arrow); b corresponds to a sub-area of 25 μm × 30 μm of Figure 3, region (2) (arrow); c corresponds to a sub-region (20 μm × 30 μm) of the micro vessel of Figure 5 (V1) in a control sample. For all areas we report the distribution of Ca, P, S, their co-localization and their XRF intensities at some selected points (table in Figure). The two structures of the MS2 sample show higher intensities of Ca and P in areas low in S. Most importantly the ratio between Ca and P XRF signals on the suspected calcification ranges between 1.7 and 1.92. On the contrary the control sample shows a greatly reduced Ca concentration and the Ca/P XRF intensity ratio is always below 1. Although the results do not allow an absolute quantification, from the counts obtained at 4.12 keV energy on selected points (1 μm × 1 μm) and reported in the table of Figure 6, one can see that in MS samples (panels a and b) the Ca values are from 5 to 8 times higher than those in the control selections (panel c). The approximate estimations in terms of ppm obtained from the analyses at 12.74 keV of the same points (average over an area of 10 μm × 10 μm) seem to reveal a much greater difference. However, since the GeoPIXE software makes a statistical approximation of a larger area which includes pixels having Ca concentration below the detection limit, the real concentration in the control samples could be underestimated. Moreover the incident energy used at XFM beamline, 12.74 keV, is not ideal for detecting low concentration of Ca, while the 4.12 keV used at beamline ID21 allows more efficient calcium detection. In table we also report signal intensities recorded at this energy for Mg. Although at this energy Mg fluorescence values are reduced by the not optimal detection efficiency, it is noted that the elemental distribution is clearly correlated to that of Ca and P. Statistical correlation analyses were performed on more than 20 points collected on calcifications and indicate highly significant positive correlation for Ca and P (Pearson r: 0.9965; p ≤ 0.001), for Ca and Mg (Pearson r: 0.9646; p ≤ 0.002) and significant negative correlation for Ca and S (Pearson r: −0.8515; p ≤ 0.05). Note that a correlation exists, although less significant, in control samples (around 20 randomly collected points) for Ca and P (Pearson r: 0.981; p < 0.1), while no significant correlation is found for Ca and S (p > 0.5), These results are in agreement with the co-localization panels highlighting the Ca-P hotspots in MS samples present in regions very low in S.

XRF elemental maps obtained at 4.12 keV.
Upper row (a), (25 μm × 30 μm) elemental maps of S, P, Fe and Ca on a sub-region of the sample MS2 indicated with the red arrow in Figure 2 (region 1); row (b) (20 μm × 30 μm): elemental maps of S, P, Fe and Ca on a sub-region of the sample MS2 indicated with the red arrow in Figure 3 (region 2). Lower row (c) (30 μm × 15 μm): S, P, Ca and Fe elemental maps of a sub-region of the V1 control sample. The table reports the intensity (intended as peak counts) for Ca, P, S and Mg (over a 1 μm × 1 μm area); and the corresponding ppm Ca concentrations (over a 10 μm × 10 μm) centered in the points indicated in the images (calculated at 12.74). In Ca maps of panels a and b dashed gray lines delineate high counts regions in calcifications.
Synchrotron Micro-XANES analyses
In order to get some insights on the chemical nature of calcium and to possibly confirm its calcified phase, XANES measurements were collected on Ca K-edge on ~1 μm2 spots selected from the Ca maps of diseased subjects (MS2) and control (V2) samples (Figure 7, panel b). The measured spectra were deconvoluted using spectral standards for hydroxyapatite, calcium phosphate, calcium carbonate, calcium stearate, calcium sulphate (Figure 7, panel a), in order to have salts both with and without phosphorus or organic and inorganic salts. According to Athena software the representative Ca K-edge spectra shown in Figure 7b are apparently dominated by features characteristic for hydroxyapatite for MS2 samples (average composition higher than 56% in weight) while for control samples this Ca salt could account for not more than 29%. The spectra in the latter case seem to be more similar to those of Ca stearate (organic salt).

XANES spectra across Ca K edge.
Panel a show the XANES spectra of the standard samples across Ca K edge. Panel b depicts the XANES spectra collected on samples MS2 and V2, in the same points indicated with the same numbers in Figure 6. In the case of sample V2 spectra the linear combination fitting performed with the Athena software using the reference standard attributes 41–47% in composition to Ca Stearate, 24–29% to Hydroxyapatite, 6–7% Ca Carbonate and 17–28% Ca Sulphate. For MS samples the fitting gives different compositions: 0–19% Ca Stearate, 56–64% to Hydroxyapatite, 0–9% Ca Carbonate and 19–27% Ca Sulphate.
Discussion
The IJV flow objectively measured by means of 2D MR drains approximately 75% of the cerebral arterial inflow, whereas it falls in CCSVI condition to respectively 64% and 52% if one or both IJVs are striken by TVMs21. The extensive flow diversion through collaterals channels permits either to avoid intra-cranial hypertension or to drain the brain22.
However, reduced perfusion and cerebro-spinal fluid flow represent the two main consequences of CCSVI on brain pathophysiology, as highlighted by the means of objective non conventional MRI measures11,12,13,14,15.
The interest in understanding more about the association of impaired cerebral venous drainage with MS and several neurodegenerative disorders7,8,9,10, lead us to investigate the jugular wall affected by TVMs characteristic of the CCSVI condition.
In order to unravel potential biochemical changes in the jugular vein wall of CCSVI patients associated to MS patients, in the present work we have combined XRF analyses with different synchrotron set-ups, spatial resolution and energies on the same tissue sections. The use of different experimental stations allowed us to acquire complementary data and to highlight the chemical differences of normal tissues vs. those with the TVMs pathology, given that each set up has a specific application field in term of spatial resolution and accessible chemical elements.
Since the analyses are only related to two patients, this explorative study has the main aim of testing the possibility of evaluating the presence of morphological alterations in the vein walls suggested by the histological examinations. For both TVMs and control samples, in fact all XRF analyses of tissue sections were coupled with the observation of conventionally stained consecutive tissue sections. The histological examinations of the two patient tissues revealed the presence of rare and small basophilic bodies suggestive of micro-calcifications within the connective tissue of the tunica adventitia. Interestingly, these structures were observed associated with the lumen of the vasa vasorum and their frequency was greatly related to the presence of these microvessels in the tissue sections. Histological staining methods, which are commonly used to visualize tissue calcifications (von Kossa and Alizarin red), were ineffective on our samples suggesting that they may be not sensitive enough to reveal small calcium precipitates, as already reported for small calcium granules at pre-artheroma stages in arteriosclerosis samples [36]. The XRF analyses reported here confirm the presence of unusual calcifications particularly on the walls of the vasa venarum of TVMs patients. The rapid analyses with the Maia detector enabled the examination of a good number of consecutive tissue slices of MS and control samples. By mapping Fe, Zn and Ca it was possible to recognize tissue structures in the unstained samples and compare them with the stained reference. By carefully observing the maps and resolving them in sub-areas, we noted a clear change in Ca signal in relationship to the vasa venorum in the samples. When comparing microvessels from the two MS patients to those from the control samples with the XRF set-up at 12.74 keV, it is evident that the first samples have more Ca in the wall. These results were confirmed in several regions of different tissue slices of both MS1 and MS2 samples. The large increase of calcium in the thin endothelium of TVMs microvessels clearly suggests an altered metabolism of this element in the tissue.
The observation is clarified by the analyses performed at lower energies (7.2, 4 and 1.5 keV) allowing the detection of P and S together with Fe and Ca and revealing also the distribution of lighter elements such as Mg, Na, C and O.
As shown in Figures 3 and 4, the MS jugular tissues clearly reveal small calcium containing structures), possibly phosphate salts, with traces of Mg, that are found in tissue regions poor in S, O and C. The main difference of MS samples versus the controls consists in the signal intensity and distribution of Ca, particularly when compared to the other elements. The Ca containing structures in MS samples are clearly compatible with calcifications. Although this is the first work suggesting the presence of small calcifications in a vein wall (and capillaries), the elemental distributions and concentrations resemble those found by other authors in artery micro-calcifications of human and animal models, obtained with similar advanced analytical techniques (such as PIXE)20,23,24. In fact, these authors report the presence of intimal micro-calcifications ranging from 6 μm to 70 μm in diameter, clearly identified by PIXE through high abundance of Ca and P together with absence of S and C. The decreased C, S and O signals in the presence of Ca deposits may be due to the low concentration of these elements compared with the rest of the tissue or to the attenuation of these low energy signals because of the calcification.
As previously mentioned, with ID21 and TwinMic set-ups absolute quantification was not performed. Nevertheless, since the analyses were carried out on tissues with the same thickness and the same experimental conditions (flux intensity, acquisition time, spot size, geometrical parameters), the results can be compared to highlight relative elemental concentrations and ratios. Calcifications are a frequent event in many pathologies and consist of a great variety of amorphous or crystalline calcium phosphates. Chemical microanalyses performed on these formations by various analytical techniques, that are able to precisely quantify the concentration of Ca and P, allow for the calculation of the calcium to phosphorus ratio23. This parameter is used to better assess the possible nature of the phosphate salts and stage of mineralization. Since these formations are usually composed of a mixture of different salts, the reported Ca/P atomic ratios range from 1.29 (indicative of dicalcium phosphate dehydrate) to 2.16 (indicative of hydroxyapatite). In our analyses we cannot compare precise quantities, particularly because P fluorescence reabsorption is greatly affected by the matrix composition and density, parameters that are very hard to estimate in our samples. Nevertheless ratios of fluorescence signal intensities of elements can be compared to distinguish different phases. It can be noted that in Figures 3 and 4 the calcium to phosphorus XRF intensity ratio on the microvessel lumen as well as in the suspected calcification is greater than 1.5. On the contrary, it is clearly lower than 1 on the lumens of microvessels in control samples. In this last case Ca and P signals simply reflects the normal distribution of the two elements in the cellular structures of these regions.
The difference is better evidenced in the analyses performed at 4.12 keV. At this energy, in fact, there is an increase in fluorescence emission efficiency for Ca, P and S. As reported in Figure 6 (table), the X-ray fluorescence counts ratio between calcium and phosphorus ranges from 1.7 to 1.92 in the Ca hotspots with signal above 1000 counts, (panels a and b). In contrast, when the Ca signal is below this value, as in all the selected points of the control samples (panel c), the ratio is always below 1. Our analyses cannot precisely identify the chemical nature of the Ca precipitates at this stage. However the observed different ranges of the Ca/P XRF intensity ratio in the diseased tissues are a strong suggestion of microcalcifications, probably similar to those reported in the arteries during early or advanced arteriosclerosis. This is further supported by the XANES analyses (Figure 7). In control tissues, the major contributions in the XANES spectra seem to come from organic Ca salts. On the contrary, in the diseased subject tissues the XANES results are in line with a substantial presence of hydroxyapatite (and other inorganic calcium salts) in clear connection with the vasa venorum. Nevertheless, the complex nature of these calcium precipitates requires further analyses (possibly in conjunction with other techniques) to definitely resolve the different Ca phases.
Besides the description of the association between extracranial TVMs of the cerebral veins and neurodegenerative disorders4,5,6,7,8,9,10, little is known about the jugular wall, since this is a new entity in vascular pathology. In a post mortem study comparing MS patients with people who died for different reasons, valvular and other intraluminal abnormalities were identified in 72% of MS patients and in 17% of controls. In contrast, vein wall stenosis occurred with similar frequency in both groups25. Coen et al. also demonstrated that in the jugular vein wall of CCSVI patients associated to MS patients there is not an increased T cell infiltration with respect to control tissue. This seems to exclude the hypothesis that venous abnormalities are a product of MS immune reaction25. The results of our exploratory study cannot reveal if the abnormal calcium deposition in jugular tissue is related to the pathogenetic mechanisms of MS and/or if it is a collateral manifestation linked to the CCSVI condition in the two patients investigated.
However, our findings clearly constitute a novel observation for microvessels and vein tissues in general, that if confirmed in a larger group of patients, could contribute to further understanding of CCSVI condition.
Altered haemodynamic forces and turbulences described in CCSVI act either on the endothelial cells or on the deeper layers of the vein wall, increasing the mechanical stress on the adventitial intramural vessels5,7,8,9,12,14,22. The mechanical overload may potentially lead to the differences demonstrated in calcium contents with respect to the control tissue. From this point of view calcium deposits could be interpreted as the result of chronic stress of the vein wall capillary with mechanical injury and damage26,27.
Finally, the results we show in this work demonstrate the advantage of applying advanced synchrotron microspectroscopy techniques, particularly consecutively at different and complementary energies and spatial resolutions, to explore histological changes that cannot be completely resolved by conventional analyses and that could suggest important pathological mechanisms.
Methods
Patients and tissues
Human samples were obtained from two patients both affected by secondary progressive multiple sclerosis associated with CCSVI and admitted to the Department of Surgery at the St. Anna Hospital, Ferrara, Italy. Tissue analysis for research on waste specimens harvested in the course of vascular procedures was approved by the local Ethical Committee (University-Hospital of Ferrara). The procedures were in accordance with the Declaration of Helsinki and all participant subjects gave written informed consent.
The first case (MS-1) was a 53 y.o. female, with 20 years disease duration and EDSS disability score 5.5. She was within normal range of thyroid hormones, normal serum calcium and vitamin D levels, normal range of serum iron and ferritin levels. Over the years during the illness, she refused any proposal of immunomodulatory/suppressant therapy. She took supplements regularly and especially vitamin D and calcium. She was admitted to the hospital for thyroid cancer. Preoperative assessment demonstrated truncular venous malformation of the left internal jugular vein by echo-color Doppler investigation and magnetic resonance of the neck. Reflux and obstruction of the flow in the lower segment of the vein were accompanied by abundant substituted circles of the thyroid vein. Since thyroidectomy would have disrupted the venous drainage, we decided to repair during the procedure the jugular steno-obstruction by an autologous saphenous patch angioplasty. A tissue specimen of the malformed and hypoplasic jugular wall was harvested for further investigation.
The second case (MS-2) was a 32 y.o. female, with 12 years disease duration and EDSS disability score 5.5. She was within normal range of thyroid hormones, serum calcium and vitamin D levels, normal range of serum iron and ferritin levels. She was treated with Copaxon, Betaferon and Mitoxantron. She was admitted to the hospital for thrombosis of the left internal jugular vein, where CCSVI was detected by color Doppler investigation six months before. At that time, the haemodynamic was characterized by significant stasis with no Doppler detectable flow due to fixed valve cusps at the junction with the brachio-cephalic trunk. Emergent intervention was aimed to prevent any embolization from a fresh thrombus found just at the malformed valvular level. Jugular tissue sample was collected during the open operation.
Out of 42 archival harvested samples analyzed by conventional histology, we selected two control tissues reassembling more the MS samples in region origin and endothelial side exposition. They were obtained from the internal jugular vein wall of healthy subjects (V1 and V2), who underwent vein repair for traumatic reasons. Particularly, both controls were tissue samples of the junctional segment of the internal jugular vein. Consequently, the selected control tissues were more comparable to the patients' tissue. All the tissues, fixed in formalin, were processed for routine paraffin embedding. Several transverse sections (10 μm thick) perpendicular to the longitudinal axis of the vessel were cut with a microtome (Leica, Italy) at approximately 0.5 μm intervals. Sections were alternately deposited on a glass slide for histological control or on Ultralene foils (Spex-Certiprep, 4 μm thick) for XRF microspectroscopy. Sections for microspectroscopy were simply dried at room temperature. Sections for histology were deparaffinized and stained with haematoxylin and eosin (HH) for morphological and general inspection, von Kossa impregnation for mineral deposits and Alizarin red for calcium depositions. After staining, samples were analyzed using a Leica Microscope (Leica Microsystems GmbH, Germany). Chemicals were purchased from Sigma-Aldrich. Scanning Electron Microscopy procedures, performed as previously reported [19], are detailed in the Supplementary Information (Material and Methods).
Synchrotron XRF elemental mapping
The synchrotron XRF experiments were carried out in three different synchrotron facilities. In all the three experimental stations the X-ray beam was focused on the samples through suitable X-ray optics. The samples were raster-scanned across the X-ray probe and in each pixel of the raster scan the emitted X-ray Fluorescence photons were collected by energy dispersive silicon detectors.
The analyses were first performed at the XFM beamline28 of the Australian Synchrotron (Clayton, Melbourne, Victoria, Australia) where the monochromatic 12.74 keV X-ray beam delivered by a Si(111) monochromator was focused by Kirkpatrick-Baez (KB) mirrors to a spot size of 2 μm × 2 μm. The XRF photons emitted by the sample were collected by the 384-element Maia detector29,30. At the XFM beamline the incident X-ray beam passes through the axis of the detector from the rear, hits the sample and fluorescence in backward direction is collected by the detector. The sample plane is 10 mm from the detector wafer and each detector element has approximately 1 mm2 active area. The large solid angle subtended by this detector coupled with fast event mode data acquisition enables dwell times of the order of hundreds μs/pixel providing megapixel elemental maps in an hour scan time31. For our sample acquisition times of 3–15 ms/pixel were used to ensure good statistics and the sample was raster-scanned with a 2 μm step size. Elemental concentrations were quantified by measuring standard thin foils of Pt, Fe and Mn of known areal concentration and thickness.
Smaller areas of the same tissue sections were then analysed at the ID21 beamline32,33 of the ESRF synchrotron facility (Grenoble, France). The sample was excited with a monochromatic incident X-ray beam of 7.2 keV (above the Fe K-edge) or 4.12 keV (above the Ca K edge) provided by the Si (111) double-crystal monochromator. The beam was focused on the sample through a zone plate diffractive optics delivering a spot size of 0.2 μm × 0.9 μm at the sample position. The sample was mounted on a custom x-y-z stage, tilted by 30° from the optical axis. A silicon drift diode detector (Bruker, Germany) with 80 mm2 active surface equipped with a low photon energy transmitting polymer window was used to collect the XRF photons emitted by the specimen. For the elemental mapping a dwell time of 300 ms per pixel and a step size of 0.5 μm was used.
Higher resolution images and elemental maps at low X-ray energy (1.5 keV) were finally collected on selected areas at the TwinMic beamline34 of the Elettra synchrotron facility (Trieste, Italy). A beam size of 0.5 μm diameter delivered by zone plate optics was used and the XRF spectra were collected by 8 Silicon Drift Detectors (SDDs) arranged in an annular geometry35,36. The sample was raster-canned with a step size of 0.5 μm. Simultaneously absorption and phase contrast images were acquired by a fast readout CCD camera37. Due to the low fluorescence yield at 1.5 keV a dwell time of 10 s/pixel was necessary in order to collect a good statistic. The TwinMic measurements were performed in vacuum (at a pressure of 10−6 mbar) to avoid significant attenuation of the fluorescence signal.
Data analysis of XRF spectra and elemental images was performed using GeoPIXE software for XFM data38 and with PyMCA software39 for ID21 and TwinMic data.
Synchrotron XANES measurements
The micro-XANES analyses were carried out at the ID 21 beamline at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The X-ray beam energy was scanned between 4.02 and 4.14 keV with 0.2 eV energy steps. Each XANES spectrum was acquired as the sum of five quick scans with 500 ms integration time per point, for a total integration time of 2.5 s per point, to monitor possible photo-reduction effects linked to the beam. No modification of the XANES spectra with exposure time was observed. XANES spectra of reference compounds (hydroxyapatite, calcium phosphate, calcium carbonate, calcium stearate, calcium sulphate) were collected to allow identification of the phases present in the sample by linear combination fitting. The spectra were base line subtracted and normalized to unity prior to fitting with the Athena software package40.
References
Lee, B. B. et al. Diagnosis and Treatment of Venous Malformations Consensus Document of the International Union of Phlebology (IUP): updated 2013. Int. Angiol. (2014).
Lee, B. B. et al. Primary Budd-Chiari syndrome: outcome of endovascular management for suprahepatic venous obstruction. J. Vasc. Surg. 43, 101–108 (2006).
Zamboni, P. et al. Membranous obstruction of the inferior vena cava and Budd-Chiari syndrome. Report of a case. J. Cardiovasc. Surg. (Torino) 37, 583–587 (1996).
Laupacis, A. et al. Association between chronic cerebrospinal venous insufficiency and multiple sclerosis: a meta-analysis. CMAJ. 183, E1203–E1212 (2011).
Zivadinov, R. et al. Chronic cerebrospinal venous insufficiency in multiple sclerosis: diagnostic, pathogenetic, clinical and treatment perspectives. Expert. Rev Neurother. 11, 1277–1294 (2011).
Zwischenberger, B. A., Beasley, M. M., Davenport, D. L. & Xenos, E. S. Meta-analysis of the correlation between chronic cerebrospinal venous insufficiency and multiple sclerosis. Vasc. Endovascular. Surg. 47, 620–624 (2013).
Chung, C. P. et al. Jugular venous reflux and white matter abnormalities in Alzheimer's disease: a pilot study. J. Alzheimers. Dis. 39, 601–609 (2014).
Di, B. F. et al. Chronic cerebrospinal venous insufficiency in Meniere disease. Phlebology. (2014).
Filipo, R. et al. Chronic cerebrospinal venous insufficiency in patients with Meniere's disease. Eur. Arch. Otorhinolaryngol. (2013).
Manju, L., haibo, X., Yuhui, W., Yi, Z. & Shuang, X. Patterns of chronic venous insufficiency in the dural sinuses and extracranial draining veins and their relationship with white matter hyperintensities for patients with Parkinson's disease. Journal of Vascular Surgery (2014).
Garaci, F. G. et al. Brain hemodynamic changes associated with chronic cerebrospinal venous insufficiency are not specific to multiple sclerosis and do not increase its severity. Radiology 265, 233–239 (2012).
Zamboni, P. et al. Hypoperfusion of brain parenchyma is associated with the severity of chronic cerebrospinal venous insufficiency in patients with multiple sclerosis: a cross-sectional preliminary report. BMC. Med. 9, 22 (2011).
D'haeseleer, M., Cambron, M., Vanopdenbosch, L. & De, K. J. Vascular aspects of multiple sclerosis. Lancet Neurol. 10, 657–666 (2011).
Zamboni, P. et al. The severity of chronic cerebrospinal venous insufficiency in patients with multiple sclerosis is related to altered cerebrospinal fluid dynamics. Funct. Neurol. 24, 133–138 (2009).
Magnano, C. et al. Cine cerebrospinal fluid imaging in multiple sclerosis. J. Magn Reson. Imaging 36, 825–834 (2012).
Ari Ide-Ektessabi Applications of synchrotron radiation: micro beams in cell micro biology and medicine. Springer Berlin Heidelberg. (2007).
Ortega, R., Deves, G. & Carmona, A. Bio-metals imaging and speciation in cells using proton and synchrotron radiation X-ray microspectroscopy. J. R. Soc. Interface 6 Suppl 5, S649–S658 (2009).
Kaulich, B. et al. Low-energy X-ray fluorescence microscopy opening new opportunities for bio-related research. J. R. Soc. Interface 6 Suppl 5, S641–S647 (2009).
Auriat, A. M. et al. Ferric iron chelation lowers brain iron levels after intracerebral hemorrhage in rats but does not improve outcome. Exp. Neurol. 234, 136–143 (2012).
Roijers, R. B. et al. Microcalcifications in early intimal lesions of atherosclerotic human coronary arteries. Am. J. Pathol. 178, 2879–2887 (2011).
Feng, W. et al. Quantitative flow measurements in the internal jugular veins of multiple sclerosis patients using magnetic resonance imaging. Rev Recent Clin. Trials 7, 117–126 (2012).
Zamboni, P. et al. An ultrasound model to calculate the brain blood outflow through collateral vessels: a pilot study. BMC. Neurol. 13, 81 (2013).
Roijers, R. B. et al. Early calcifications in human coronary arteries as determined with a proton microprobe. Anal. Chem. 80, 55–61 (2008).
Debernardi, N. et al. Microcalcifications in atherosclerotic lesion of apolipoprotein E-deficient mouse. Int. J. Exp. Pathol. 91, 485–494 (2010).
Diaconu, C. et al. A Pathologic Evaluation of Chronic Cerebrospinal Venous Insufficiency (CCSVI) (P05.125). Neurology (2013).
Tisato, V. et al. Endothelial PDGF-BB produced ex vivo correlates with relevant hemodynamic parameters in patients affected by chronic venous disease. Cytokine 63, 92–96 (2013).
Havelka, G. E. & Kibbe, M. R. The vascular adventitia: its role in the arterial injury response. Vasc. Endovascular. Surg. 45, 381–390 (2011).
Paterson, D. et al. The X-ray Fluorescence Microscopy Beamline at the Australian Synchrotron. AIP Conference Proceedings 1365, 219–222 (2011).
Howard, D. L. et al. High-definition X-ray fluorescence elemental mapping of paintings. Anal. Chem. 84, 3278–3286 (2012).
James, S. A. et al. Quantitative comparison of preparation methodologies for X-ray fluorescence microscopy of brain tissue. Anal. Bioanal. Chem. 401, 853–864 (2011).
Lombi, E., de Jonge, M. D., Donner, E., Ryan, C. G. & Paterson, D. Trends in hard x-ray fluorescence mapping: environmental applications in the age of fast detectors. Anal Bioanal Chem. 400, 1637–1644 (2011).
Szlachetko, J. et al. Wavelength-dispersive spectrometer for X-ray microfluorescence analysis at the X-ray Microscopy beamline ID21 (ESRF). J. Synchrotron. Radiat. 17, 400–408 (2010).
Bohic, S. et al. Biomedical applications of the ESRF synchrotron-based microspectroscopy platform. J. Struct. Biol. 177, 248–258 (2012).
Kaulich, A. European Twin X-ray Microscopy Station Commissioned at ELETTRA. Conf.Proc.Series IPAP 7, 22–25 (2006).
Gianoncelli, A., Morrison, G. R., Kaulich, B., Bacescu, D. & Kovac, J. A fast read-out CCD camera system for scanning X-ray microscopy. Appl. Phys. Lett 89, 251117–251119 (2006).
Gianoncelli, A. et al. Simultaneous Soft X-ray Transmission and Emission Microscopy. Nuclear Instruments and Methods in Physics Research A 608, 195–198 (2009).
Morrison, G. R., Gianoncelli, A., Kaulich, B., Bacescu, D. & Kovac, J. A fast read-out CCD system for configured-detector imaging in STXM. Conf.Proc.Series IPAP 7, 277–379 (2006).
Ryan, C. G. et al. Elemental X-ray imaging using the Maia detector array: The benefits and challenges of large solid-angle. Nucl. Instr. Meth A 619, 37–43 (2010).
Sole, A. et al. A multiplatform code for theanalysis of energy-dispersive X-rayfluorescence spectra. Spectrochim Acta B 62, 63–68 (2007).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Acknowledgements
The Authors would like to acknowledge G. Kourousias, C. Ryan for their invaluable technical support. The Authors also thanks Dr. Maria Palumba and Dr. Maria Rita Bovolenta for their help in Scanning Microscopy analyses. Part of this research was undertaken on the XFM beamline at the Australian Synchrotron, Victoria, Australia. The authors are grateful to the Australian Synchrotron for providing the beamtime and to D. Howard, M. de Jonge and K. Spiers for technical support at the XFM beamline. This study was partially supported by the Italian Ministry of Education, University and Research (MIUR Programme PRIN 2010–2011), Grant no. 2010XE5L2R. We thank Dr. K. Prince and S. Vielle for careful reading of the manuscript.
Author information
Authors and Affiliations
Contributions
L.P. and P.Z. designed and coordinated the study. L.P., A.G., C.C., V.T., M.S. and C.C. performed the experiments. L.P., A.G., C.C., V.T., F.S. and D.P. were involved in analyzing the data. M.S. and D.P. contributed analysis tools. L.P., A.G. and P.Z. wrote the paper and all the authors participated in revising. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Pascolo, L., Gianoncelli, A., Rizzardi, C. et al. Calcium micro-depositions in jugular truncular venous malformations revealed by Synchrotron-based XRF imaging. Sci Rep 4, 6540 (2014). https://doi.org/10.1038/srep06540
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06540
This article is cited by
-
Modelling of the dilated sagittal sinuses found in multiple sclerosis suggests increased wall stiffness may be a contributing factor
Scientific Reports (2022)
-
Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: a review
Cell Death & Disease (2018)
-
Physicochemical characterization of mineral deposits in human ligamenta flava
Journal of Bone and Mineral Metabolism (2018)
-
Impaired Neurovisceral Integration of Cardiovascular Modulation Contributes to Multiple Sclerosis Morbidities
Molecular Neurobiology (2017)
-
Differential protein folding and chemical changes in lung tissues exposed to asbestos or particulates
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
