
Abstract
Engineered endonucleases, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system, provide a powerful approach for genome editing in animals. However, the microinjection of endonucleases into embryos requires a high skill level, is time consuming and may cause damage to embryos. Here, we demonstrate that the electroporation of endonuclease mRNAs into intact embryos can induce editing at targeted loci and efficiently produce knockout rats. It is noteworthy that the electroporation of ZFNs resulted in an embryonic survival rate (91%) and a genome-editing rate (73%) that were more than 2-fold higher than the corresponding rates from conventional microinjection. Electroporation technology provides a simple and effective method to produce knockout animals.
Similar content being viewed by others

Introduction
The rat is an important animal model that has been used to understand human diseases1,2. Genetically modified (GM) rat strains are now being used to model human diseases in various research fields3,4. Engineered endonucleases, including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system, are invaluable tools for the rapid generation of GM animals including rats5,6,7,8. Importantly, these new technologies provide genome-editing approaches for a wide variety of organisms that were previously inaccessible without embryonic stem (ES) cells9,10,11 and induced pluripotent stem (iPS) cells12,13. GM animals are usually produced by microinjecting engineered endonucleases into pronuclear-stage embryos5,6. Although this method is now the gold standard, it requires sophisticated manual skills to prevent cell damage. Additionally, microinjection is not convenient when many cells need to be assessed simultaneously because the DNA/RNA has to be injected into embryos one by one using a micromanipulator. Electroporation is another method that introduces exogenous DNA/RNA into embryos. However, the current protocols require that the zona pellucida of the embryos is weakened by treatment with Tyrode's acid solution before electroporation for the efficient introduction of DNA14,15. To simplify these procedures, we introduced ZFN, TALEN, or CRISPR/Cas mRNA into intact rat embryos without weakening the zona pellucida by electroporation using the Super Electroporator NEPA 21 (NEPA GENE Co. Ltd., Chiba, Japan).
Results
Introduction of mRNA using an electroporator
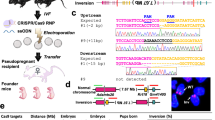
ZFN, TALEN, or CRISPR mRNA was electroporated into embryos (Fig. 1a). Pronuclear-stage embryos were placed in a line on the glass chamber between metal plates that were filled with phosphate-buffered saline (PBS) containing mRNA (Fig. 1a). mRNAs were efficiently introduced into intact embryos with a 3-step pulse system (Fig. 1b). In the first step, the poring pulse makes micro-holes in the zona pellucida and oolemma. In the second step, several of the first transfer pulses transfer mRNA into the cytoplasm. In the third step, the polarity-changed second transfer pulse increases the chance of transferring mRNA into embryos (Fig. 1c).

Introducing mRNA into embryos by electroporation.
(a) Super Electroporator NEPA 21 (left) and glass chamber with metal plates (right). Embryos were placed in a line between the metal plates (arrow). (b) Illustration of electric pulses delivered by the electroporator. (c) The first poring pulse with high voltage and short duration makes micro-holes in the zona pellucida and oolemma (left) and mRNA in PBS was then moved into the cytoplasm with a few first transfer pulses with a low voltage and long duration after the poring pulse (middle). The mRNA was then transferred into oocytes by the polarity-changed second transfer pulse (right). (d) Fluorescence analysis of tetramethylrhodamine-labelled dextran that was introduced into embryos by electroporation with the pulse width adjusted to 0, 0.5, 1.5 and 2.5 ms.
Introduction of tetramethylrhodamine-labelled dextran into intact pronuclear-stage embryos
To test the electroporation conditions to introduce materials into embryos, tetramethylrhodamine-labelled dextran (3 kDa molecular weight), which is easily visualised and nontoxic to living cells, was used. Pronuclear-stage embryos were collected from superovulated female rats the day after mating. Tetramethylrhodamine-labelled dextran was electroporated into intact rat embryos using the electroporator with the pulse width adjusted to 0, 0.5, 1.5 and 2.5 ms. Dextran was located in the whole cytoplasm of the embryos (Fig. 1d).
Introduction of the ZFN or TALEN plasmid into fibroblast-like cells
ZFN and TALEN plasmids were designed to target exon 2 of the rat interleukin 2 receptor gamma (Il2rg) gene (Supplementary Fig. S1)16,17. To evaluate the activity of the ZFN and TALEN, we electroporated these expression vectors into rat fibroblast-like (Rat-1) cells. After 1 week, the cells were collected and the genomic DNA was extracted and analysed for mutations using the Surveyor (Cel-1) nuclease assay. The Rat-1 cells that were transfected with the plasmids showed a cleavage activity of 2.6% for ZFN and 29.3% for TALEN compared with the negative control cells that were transfected with the GFP expression vector (Supplementary Fig. S2). Sequence analysis of the Il2rg locus also revealed mutation rates of 10.4% (10 mutations per 96 colonies screened) for ZFN and 19.8% (17 mutations per 86 colonies) for TALEN (Supplementary Fig. S3).
Introduction of ZFN or TALEN mRNA into intact pronuclear-stage embryos
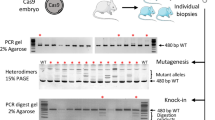
ZFN or TALEN mRNA was electroporated into intact rat pronuclear-stage embryos. The ZFN or TALEN mRNA was transcribed from the plasmids; mRNA concentrations of 40 μg/mL and 10 μg/mL were used for electroporation and microinjection, respectively. The transcribed mRNA was electroporated into intact embryos with a poring pulse (voltage: 225 V; pulse interval: 50 ms; pulse width: 0.5, 1.5 and 2.5 ms; number of pulses: 4). After electroporation, the embryos that developed to the 2-cell stage were transferred into the oviducts of pseudopregnant females. Of the embryos that were microinjected with ZFN mRNA, which served as a control in this study, 10% developed into offspring and 33% of the offspring had an edited Il2rg locus (Table 1). In contrast, 31, 24 and 6% of the embryos that were electroporated with the pulse width adjusted to 0.5, 1.5 and 2.5 ms, respectively, developed into offspring. Of these offspring, 37, 73 and 75%, respectively, had an edited Il2rg locus (Table 1, Supplementary Fig. S4). The germline transmission of the ZFN electroporation-induced editing was also confirmed in the next generation (Supplementary Table S1).
Of the embryos that were microinjected with TALEN mRNA, 12% developed into offspring and all 6 of the delivered pups had an edited Il2rg locus (Table 1). TALEN mRNA was electroporated into intact pronuclear-stage embryos with the same poring pulse that was used for the ZFN electroporation and the pulse width was adjusted to 1.5 and 2.5 ms. Of the embryos that were electroporated with TALEN mRNA with the pulse width adjusted to 1.5 and 2.5 ms, 44 and 30%, respectively, developed into offspring and 4 and 18%, respectively, had an edited Il2rg locus (Table 1, Supplementary Fig. S5).
Introduction of Cas9 mRNA and guide RNA into intact pronuclear-stage embryos
Cas9 mRNA and single-stranded guide RNA (gRNA) targeting the rat Il2rg gene (Supplementary Fig. S1) were electroporated into intact rat pronuclear-stage embryos with a poring pulse (voltage: 225 V; pulse interval: 50 ms; pulse width: 1.5 and 2.5 ms; number of pulses: 4). Cas9 mRNA and gRNA were used at concentrations of 2,000 and 1,000 μg/mL for electroporation and 100 and 50 μg/mL for microinjection, respectively. After the electroporation or microinjection, the embryos that developed to the 2-cell stage were transferred into the oviducts of pseudopregnant females. Of the embryos that were microinjected with Cas9 mRNA and gRNA, which served as a control in this study, 41% developed into offspring and 51% of the offspring had an edited Il2rg locus (Table 1, Supplementary Fig. S6). In contrast, 43 and 55% of the embryos that were electroporated with the pulse width adjusted to 1.5 and 2.5 ms, respectively, developed into offspring. Finally, 9% of the offspring derived from embryos that were electroporated with a pulse width adjusted to 2.5 ms had an edited Il2rg locus (Table 1, Supplementary Fig. S6).
Discussion
Here, we established a method to produce knockout rats by introducing ZFN, TALEN and CRISPR/Cas mRNA into intact embryos using electroporation. Introducing DNA/RNA into embryos by electroporation has been used to analyse the mechanism of embryo development and expression of DNA/RNA in preimplantation embryos in vitro. However, previous methods required that the zona pellucida was weakened with Tyrode's solution before electroporation14,15. The weakening of the zona pellucida may affect the subsequent embryonic development because of its importance in embryonic development in vivo18,19. We observed that tetramethylrhodamine-labelled dextran was present in the whole cytoplasm of the intact embryos after electroporation (Fig. 1d). Our method using the Super Electroporator NEPA 21 introduced satisfactory nucleases even if intact embryos were used. This method was simple and effective compared with the conventional electroporation methods that use embryos with a weakened or removed zona pellucida (Table 1).
In general, knockout animals have been produced by injecting targeted genes that were modified by homologous recombination into blastocysts of ES cells11. This process requires sophisticated manual skills and the success rates of knockout animals varied in each strain20. Engineered endonucleases have been used to edit targeted genes in nematodes, flies, zebrafish, frogs and human ES cells and iPS cells21,22. It was also reported that these mRNAs are highly active in rat embryos5,6,16,23. Engineered endonucleases make it possible to produce knockout animals even in strains and species that do not have established ES cell lines.
The electroporation method used in this study efficiently introduced ZFN, TALEN and CRISPR mRNA into intact embryos with a 3-step pulse system (Fig. 1b). It was thought that this electroporation system could efficiently introduce mRNA into intact embryos by using two operating transfer pulses (Fig. 1c). The introduction of nucleases to embryos that have not been treated to weaken or remove the zona pellucida possibly reduces the time necessary for embryo manipulation, facilitating the healthy development of offspring after embryo transfer. In fact, electroporation with the 3-step pulse system drastically increased the survival rate of embryos that received ZFN (91% with the pulse width adjusted to 1.5 ms) compared with that from microinjection (44%) (Table 1). Importantly, the genome-editing rate was also significantly higher with the electroporation of ZFN (73%) than with microinjection (33%), indicating the efficient introduction of mRNA by the 3-step pulse system. In the case of TALEN and CRISPR, the survival rate was increased (98 and 91% with the pulse width adjusted to 2.5 ms, respectively), but the editing rate was not (18 and 9%) (Table 1). The size of the TALEN and Cas9 mRNA, which is three times larger than that of the ZFN mRNA, might hamper its penetration through the zona pellucida. Although the editing rate of TALEN and CRISPR is sufficient for genome editing, further modifications of the electroporation conditions or an improved method that increases genome editing with the co-introduction of exonuclease 1 (Exo1) mRNA24 might be required. Further modifications should lead to increasing success rates of knockout animals.
In conclusion, we established a novel, efficient method for generating knockout rats that we called the “Technique for Animal Knockout system by Electroporation (TAKE)”. The electroporation protocol has high success rates without requiring special technical skills and it is much faster than the microinjection of multiple cells. It is also applicable to embryos from various animal species. We believe that the TAKE method will facilitate the production of genetically engineered animals to be used to study gene functions and human diseases.
Methods
Animals
All animal care and procedures that were performed in this study conformed to the Guidelines for Animal Experiments of Kyoto University and they were approved by the Animal Research Committee of Kyoto University. The F344/Stm rats used in this study were supplied by the National BioResource Project-Rat (Kyoto, Japan, http://www.anim.med.kyoto-u.ac.jp/NBR). Males that were older than 11 weeks and females that were 8 to 16 weeks old were used as sperm and oocyte donors, respectively. Jcl:Wistar females that were 8 to 16 weeks old were purchased from CLEA Japan Inc. (Tokyo, Japan) and were used as recipients for embryo transfer. All animals were maintained in an air-conditioned (temperature 24 ± 2°C, humidity 50 ± 10%) and light-controlled room (lights on from 07:00 to 19:00).
Preparation of ZFN, TALEN and CRISPR mRNA
Custom-designed ZFN plasmids from Sigma-Aldrich (St. Louis, MO, USA)23 and self-made TALEN and CRISPR plasmids were used. The protocol for TALEN assembly was previously described17. Plasmids expressing Cas9 (hCas9: ID#41815) and gRNA (gRNA_Cloning Vector: ID#41824) were obtained from Addgene (www.addgene.org/CRISPR). We designed the ZFN, TALEN and CRISPR to target the rat Il2rg gene (Supplementary Fig. S1). ZFN, TALEN and Cas9 mRNA were transcribed in vitro using a MessageMax™ T7 mRNA transcription kit (Cambio, Cambridge, UK) and polyadenylated using an A-Plus™ Poly(A) polymerase tailing kit (Epicentre Biotechnologies, Madison, WI, USA). gRNA was transcribed with a MEGAshortscript T7 Kit (Life Technologies Co., Carlsbad, CA, USA). RNA was then purified using a MEGAClear™ kit (Life Technologies Co.). mRNA of ZFNs and TALENs was resuspended in PBS at 40 μg/mL for electroporation and 10 μg/mL for microinjection. Cas9 mRNA and gRNA were resuspended in PBS at 2,000 and 1,000 μg/mL for electroporation and 100 and 50 μg/mL for microinjection, respectively.
Cell culture and transfection
Rat-1 cells were obtained from the RIKEN BRC Cell Bank (Tsukuba, Japan, http://www.brc.riken.jp/lab/cell/english). The Rat-1 cells were cultured in Dulbecco's modified Eagle's medium (Life Technologies Co.) that was supplemented with 10% fetal bovine serum (Thermo Fisher Scientific Inc., Waltham, MA, USA) in a humidified atmosphere containing 5% CO2 at 37°C. The cells (1 × 106) were suspended in 100 μL PBS, given 10 μg plasmid and electroporated with the Super Electroporator NEPA 21 (NEPA GENE Co. Ltd., Chiba, Japan) under the following conditions: (A) pulse voltage, 275 V; pulse interval, 50 ms; pulse width, 2.5 ms; and pulse number, 2; or (B) pulse voltage, 275 V; pulse interval, 50 ms; pulse width, 1.0 ms; and pulse number, 3. The in vitro transfer experiment was replicated four times.
Collection of embryos
Females were induced to superovulate by an intraperitoneal injection of 150 IU/kg pregnant mare serum gonadotropin (ASKA Pharmaceutical Co. Ltd., Tokyo, Japan) followed by an injection of 75 IU/kg human chorionic gonadotropin (ASKA Pharmaceutical Co. Ltd.) 48 h later. Females were then mated with males of the same strain overnight. Pronuclear-stage embryos were collected from oviducts of females the day after mating and they were kept in modified Krebs-Ringer bicarbonate (mKRB)25 before electroporation.
Introduction of ZFN, TALEN and CRISPR mRNA into intact embryos by electroporation
Tetramethylrhodamine-labelled dextran (3 kDa; Life Technologies Co.) that was diluted in PBS to a concentration of 2 mg/mL was introduced to observe the localisation of external material in the embryos after electroporation. Pronuclear-stage embryos were placed in a line on the glass chamber between metal plates that were filled with PBS containing tetramethylrhodamine-labelled dextran (Fig. 1a). The poring pulse (voltage: 225 V, pulse interval: 50 ms, number of pulses: 4) was selected and the pulse width was adjusted to 0.5, 1.5 and 2.5 ms. The localisation of tetramethylrhodamine-labelled dextran was observed in all embryos in each group using an inverted microscope with fluorescence (Olympus Co., Tokyo, Japan). Embryos that were exposed to PBS with tetramethylrhodamine-labelled dextran without electroporation were used as a control (Fig. 1d).
ZFN, TALEN, or CRISPR mRNA was introduced into embryos using the same method as that used to introduce tetramethylrhodamine-labelled dextran. Pronuclear-stage embryos were electroporated with the same poring pulse that was used for the tetramethylrhodamine-labelled dextran and different pulse widths (0.5, 1.5 and 2.5 ms) were used for the PBS containing ZFN mRNA. TALEN or Cas9 mRNA and gRNA were electroporated into embryos with the same poring pulse and different pulse widths (1.5 and 2.5 ms). Embryos that received microinjected mRNA in the pronucleus were used as controls. All embryos in each group were cultured in mKRB medium at 37°C under 5% CO2 and 95% air.
Embryo transfer
Embryos that developed to the 2-cell stage after the introduction of ZFN, TALEN, or CRISPR mRNA were transferred into the oviducts of surrogate females that were mated with vasectomised males the day before the embryo transfer. The number of offspring that were born naturally was counted at 21 days of gestation.
Analysis of gene editing in offspring
Editing of the targeted gene was analysed using genomic DNA that was extracted from blood that was adhered to FTA cards using a GENEXTRACTOR TA-100 automatic DNA purification system (Takara Bio Inc., Shiga, Japan). PCR was performed in a total volume of 15 μL under the following conditions: 1 cycle of 94°C for 3 min; 35 cycles of 94°C for 30 s, 60°C for 1 min and 72°C for 45 s; and 1 cycle of 72°C for 3 min. The final reaction mixture contained 100 ng genomic DNA, 200 μM dNTPs, 1.0 mM MgCl2, 0.66 μM each primer and 0.4 U Taq DNA polymerase (Life Technologies Co.). To edit the cleavage site in the genome at the Il2rg locus, two primer sets were designed to amplify small (292 bp) and large (3,158 bp) fragments as shown in Supplementary Fig. S1. The PCR products were directly sequenced using the BigDye terminator v3.1 cycle sequencing mix and the standard protocol for an Applied Biosystems 3130 DNA Sequencer (Life Technologies Co.).
Analysis of germ-line transmission
Some offspring with deletions of the targeted gene that were derived from embryos electroporated with ZFN mRNA using a pulse width of 1.5 ms were selected. They mated naturally with wildtype males or females after they had matured. We analysed the edited gene in the offspring of the next generation.
References
Jacob, H. Functional genomics and rat models. Genome Res. 9, 1013–1016 (1999).
Aitman, T. J. et al. Progress and prospects in rat genetics: a community view. Nat. Genet. 40, 516–522 (2008).
Mullins, J. J., Peters, J. & Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature 344, 541–544 (1990).
Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C. & Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Res. 5, 223–234 (1996).
Geurts, A. M. et al. Knockout rats produced via embryo pronuclear microinjection of designed zinc finger nucleases. Science 325, 433 (2009).
Tesson, L. et al. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 29, 695–696 (2011).
Li, D. et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 31, 681–683 (2013).
Li, W., Teng, F., Li, T. & Zhou, Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat. Biotechnol. 31, 684–686 (2013).
Li, P. et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell 135, 1299–1310 (2008).
Buehr, M. et al. Capture of authentic embryonic stem cells from rat blastocysts. Cell 135, 1287–1298 (2008).
Tong, C., Li, P., Wu, N. L., Yan, Y. & Ying, Q. L. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 467, 211–213 (2010).
Liao, J. et al. Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell 4, 11–15 (2009).
Li, W. et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell 4, 16–19 (2009).
Grabarek, J. B., Plusa, B., Glover, D. M. & Zernicka-Goetz, M. Efficient delivery of dsRNA into zona-enclosed mouse oocytes and preimplantation embryos by electroporation. Genesis 32, 269–276 (2002).
Peng, H., Wu, Y. & Zhang, Y. Efficient delivery of DNA and morpholinos into mouse preimplantation embryos by electroporation. PLoS One 7, e43748 (2012).
Mashimo, T. et al. Generation and characterization of severe combined immunodeficiency rats. Cell Rep. 2, 685–694 (2012).
Sakuma, T. et al. Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity. Sci. Rep. 3, 3379 (2013).
Bronson, R. A. & McLaren, A. Transfer to the mouse oviduct of eggs with and without the zona pellucida. J. Reprod. Fertil. 22, 129–137 (1970).
Modliński, J. A. The role of the zona pellucida in the development of mouse eggs in vivo. J. Embryol. Exp. Morphol. 23, 539–547 (1970).
Nagy, A., Gertsenstein, M., Vintersten, K. & Behringer, R. [Production of chimeras]. Manipulating the mouse embryo. A laboratory manual. 3rd edition. [Nagy A., Gertsenstein M., Vintersten K., & Behringer R. (eds.) [453–506] (Cold Spring Harbor Laboratory Press, 2003).
Hockemeyer, D. et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857 (2009).
Sakuma, T. & Woltjen, K. Nuclease-mediated genome editing: At the front-line of functional genomics technology. Dev. Growth Differ. 56, 2–13 (2014).
Mashimo, T. et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 5, e8870 (2010).
Mashimo, T. et al. Efficient gene targeting by TAL effector nucleases coinjected with exonucleases in zygotes. Sci. Rep. 3, 1253 (2013).
Toyoda, Y. & Chang, M. C. Fertilization of rat eggs in vitro by epididymal spermatozoa and the development of eggs following transfer. J. Reprod. Fertil. 36, 9–22 (1974).
Acknowledgements
We thank Y. Kunihiro, K. Hattori and K. Yoshimi for technical support in this study.
Author information
Authors and Affiliations
Contributions
T.K. designed the work and performed the experiments. T.S. and T.Y. constructed the TALEN plasmid. T.K. and T.M. wrote the manuscript. All authors reviewed the manuscript before submission.
Ethics declarations
Competing interests
Kyoto University and NEPA GENE Co. Ltd. have applied for a patent describing this method (patent application no. 2013-209184). T.K. and T.M. are listed as inventors of the patent and will receive part of the revenue that it may generate.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kaneko, T., Sakuma, T., Yamamoto, T. et al. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Sci Rep 4, 6382 (2014). https://doi.org/10.1038/srep06382
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06382
This article is cited by
-
Successful induction of pseudopregnancy using sonic vibration in mice
Scientific Reports (2023)
-
The Mc4r gene is responsible for the development of experimentally induced testicular teratomas
Scientific Reports (2023)
-
HOXA10 promotes Gdf5 expression in articular chondrocytes
Scientific Reports (2023)
-
A novel technique for large-fragment knock-in animal production without ex vivo handling of zygotes
Scientific Reports (2023)
-
Genome centric engineering using ZFNs, TALENs and CRISPR-Cas9 systems for trait improvement and disease control in Animals
Veterinary Research Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
