
Abstract
Protein kinase B (PKB) also known as Akt is involved in many signal transduction pathways. As alterations of the PKB pathway are found in a number of human malignancies, PKB is considered an important drug target for cancer therapy. However, production of sufficient amounts of active PKB for biochemical and structural studies is very costly because of the necessity of using a higher organism expression system to obtain phosphorylated PKB. Here, we report efficient production of active PKBα using the BmNPV bacmid expression system with silkworm larvae. Following direct injection of bacmid DNA, recombinant PKBα protein was highly expressed in the fat bodies of larvae and could be purified using a GST-tag and then cleaved. A final yield of approximately 1 mg PKBα/20 larvae was recorded. Kinase assays showed that the recombinant PKBα possessed high phosphorylation activity. We further confirmed phosphorylation on the activation loop by mass spectrometric analysis. Our results indicate that the silkworm expression system is of value for preparation of active-form PKBα with phosphorylation on the activation loop. This efficient production of the active protein will facilitate further biochemical and structural studies and stimulate subsequent drug development.
Similar content being viewed by others


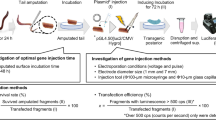
Introduction
Protein kinase B (PKB) is a well-known Ser/Thr kinase belonging to the AGC protein kinase family (protein kinase A, C and G) and plays a key role in the downstream pathways of various growth factors and hormones1. Increasing evidence indicates that PKB-mediated pathways are involved in cancer2,3,4,5. Thus, PKB is now regarded as a possible target for anti-cancer drug development. Three isoforms of PKB exist in mammals, namely PKBα, PKBβ and PKBγ (also known as Akt1, Akt2 and Akt3, respectively); all three isoforms contain an N-terminal pleckstrin homology (PH) domain, a catalytic (kinase) domain and a C-terminal regulatory segment containing a hydrophobic motif (HM)6,7.
In signal transduction pathways, the activity of PKB is controlled by allosteric changes caused by posttranslational modification. Under starvation conditions, PKB adopts an autoinhibited form and is activated by stimuli such as growth factors and hormones. To convert PKB to its active form, phosphorylation of two regulatory sites occur: Thr308 in the activation loop of PKB kinase domain and Ser473 in the hydrophobic motif of the C-terminal region (residue numbering based on human PKBα). The phosphorylation of both regulatory sites leads to the full activation of PKB8. Phosphoinositide-dependent protein kinase-1 (PDK1), another AGC protein kinase, has been shown to be responsible for phosphorylation at Thr3089,10. However, the identity of the Ser473 kinase is uncertain, although a strong candidate is Target of rapamycin complex 2 (TORC2)11,12. Effective production of active PKB is a critical problem for biochemical and structural studies of PKB and for drug development strategies such as structure-based drug design (SBDD) that target PKB. Cost effective production methods, for example, use of an E. coli expression system, are not available for achieving expression in the soluble fraction and for post translational modifications such as phosphorylation. However, the baculovirus expression vector system (BEVS) can enable production of post-translationally-modified recombinant proteins. To date, a number of structural studies on the kinase domain of PKB13,14,15,16,17 and of its complex structure with inhibitors18,19,20,21,22,23,24 have been performed using recombinant proteins derived from an Sf9 expression system. Unfortunately, the commercially available Sf9 expression systems using Autographa californica nucleopolyhedrovirus (AcNPV) BEVS have the major drawbacks of requiring large-scale culture of insect cell lines, a high-titer recombinant virus and proficiency in time-consuming virus-handling techniques. Accordingly, a cost effective technique for preparing active PKB is still awaited.
A possible route for establishing a cost effective production method for PKB is through use of a BmNPV bacmid-silkworm system25,26,27,28,29,30. This system is a combination of the Bombyx mori nuclear polyhedrosis virus (BmNPV) and a silkworm host. The BmNPV baculovirus bacmid offers some remarkable advantages: it can be directly injected into silkworm larvae; site-specific transposition to introduce the target gene into the bacmid DNA can be performed using an E. coli BmDH10Bac; and the bacmid DNA is easily prepared from E. coli BmDH10Bac culture. In addition, use of two different promoters, polyhedrin promoter and p10 promoter, allow co-expression in a single vector. Silkworm larvae are also very attractive as a host for protein production because they are simple to feed, have a low maintenance cost and provide a high level expression of the inserted expression system. Thus, large-scale and time-consuming cell culture is not required; rather, one incubator is essentially all that is needed for breeding the silkworms. To date, expressions of a number of proteins using BmNPV bacmid-silkworm system were reported (reviewed by Kato et al.)29,31, including many secretory proteins such as interferon and interleukin, trans membrane proteins and cell surface receptors such as GPCR and some intracellular proteins.
Here, we describe the successful use of the BmNPV bacmid-silkworm system to produce the recombinant kinase domain of human PKBα in a simple and cost effective manner. Notably, the PKBα was obtained in an active form with phosphorylation on the activation loop after co-expression with the activating kinase PDK1. The yield of PKBα (1 mg/20 larvae) was sufficient for biochemical and structural studies.
Results
Expression and purification of PKBα kinase domain after direct injection of bacmid DNA into larvae
The kinase domain (residues 145–481) of human PKBα was expressed using the BmNPV bacmid-silkworm system. To phosphorylate Thr308 within the activation loop, we constructed a co-expression vector, pFastGSTPKB-PDK1, that encodes PKBα fused to an N-terminal GST tag and the kinase domain of human PDK1 (Fig. 1). The bacmid DNA was prepared using E. coli BmDH10Bac and the bacmid DNA construct was directly injected into the bodies of fifth instar silkworm larvae. Five or six days after the injection, the target protein was collected from larval fat bodies, which were lysed by sonication. In a Coomassie stained SDS-PAGE gel, a visible GST-PKBα band was clearly present (Fig. 2a). The sample used here was obtained from a single larva and purified by batch GST affinity chromatography. The clear presence of a GST-PKBα band indicated a relatively high level of protein expression. For large scale preparation, 20 larvae were sampled simultaneously and their lysate applied to a GST affinity column chromatography; the protein was finally purified by size exclusion chromatography after GST tag cleavage using HRV-3C proteinase. Approximately 1 mg of highly purified protein was obtained from 20 larvae.

Construction of BmNPV bacmid vectors and application of the PKBα expression system in silkworms.
(a) Schematic representations of the domain organization of PKBα and PDK1. (b) Schematic outline of the flow chart for the BmNPV bacmid-silkworm system.

Preparation of recombinant PKB.
(a) Purification of PKBα from silkworm fat bodies. The proteins eluted from the GSH column were separated by 12.5% SDS–PAGE. The region of interest is cropped. The gray and black arrowheads indicate the GST-PKBα and PKBα proteins, respectively. (b) Size exclusion chromatography using Superdex-75(16-60). Elution profiles of PKBα and reference proteins are shown in purple and green, respectively. (c) 1D 1H NMR spectrum of PKBα.
We also constructed a bacmid that only expressed GST-fused PKB. Expression in larvae and purification of the expressed protein were performed in same manner as above. We obtained approximately 0.5–0.6 mg GST-PKB from 20 larvae. Thus, the yield of PKBα without PDK1 co-expression showed a slight decrease.
Structural integrity of the recovered PKBα
We examined the structural integrity of the PKBα obtained from the larvae. Analytical size exclusion chromatography gave an estimated molecular weight of 43 kDa by comparison with known standards (Fig. 2b). This estimate is consistent with the calculated monomeric molecular weight of 39 kDa for amino acids 145–481 of PKBα and suggests that the PKBα recovered from silkworm exists as monomer in solution. Of note, no protein was eluted in the void volume indicating that the protein was not aggregated (Fig. 2b). The 1D 1H NMR spectrum of the recovered PKBα was dispersed, especially for signals within -1 to 0.5 ppm and 8 to 10 ppm, corresponding to methyl and amide signals, respectively; the results of this analysis indicated that the PKBα had a folded structure (Fig. 2c). Overall, we concluded that the PKBα prepared from the BmNPV bacmid-silkworm system maintained structural integrity and behaved as expected in solution.
Phosphorylation assay using purified PKBα
To further characterize the PKBα produced by the BmNPV bacmid-silkworm system, we monitored its phosphorylation activity using a GSK-3 peptide fused protein as substrate. Phosphorylation was detected by Western blotting using a phospho-GSK-3α/β (Ser21/9) antibody. PKBα co-expressed with PDK1 showed a high phosphorylation activity that was slightly greater than commercially available activated PKBα (Fig. 3a). Notably, we also found that the phosphorylation activity of PKBα expressed alone was about 10 fold weaker than of PKBα co-expressed with PDK1 (Fig. 3b). This effect was evident at low concentrations. It is therefore likely that phosphorylation on the activation loop of PKBα co-expressed with PDK1 underlies its higher activity.

Phosphorylation assay.
(a) GSK3peptide fusion proteins were separated by 15% SDS–PAGE and subjected to a Western blotting analysis using a GSK3β/α antibody. Commercially available PKBα is indicated by “PKB active”, PKB with PDK1 co-expression as “PDK1+” and PKBα without PDK1 co-expression as “PDK-”. Four different panels that show the bands visualized by different exposure time are shown. (b) Western blots showing dose related variations in phosphorylation by GSK3peptide fusion proteins. (a)(b) The regions of interest are cropped.
MS analysis of the in vivo phosphorylated PKBα
To determine the phosphorylation state of the purified PKBα co-expressed with PDK1, we digested the protein with trypsin and carried out a liquid chromatography-mass spectrometric (LC-MS) analysis. In the MS spectra, we identified non- and di-phosphorylated peptides derived from the activation loop (TFCGTPEYLAPEVLEDNDYGR, residues 308–328, Fig. 4a), but were unable to detect mono-phosphorylated peptide (data not shown). In addition, the MS/MS spectrum of the di-phosphorylated peptide showed that the C-terminal residues PEYLAPEVLEDNDYGR could be confidently assigned by the presence of continuous y-series ions (y2–y14 and y16) that have no phosphorylated residues. These results suggested that the remaining N-terminal region TFCGT contained two phosphorylated residues (Fig. 5a), namely, Thr308 and Thr312.

Identification and stoichiometric quantitation of PKBα phosphorylated peptides by LC-MS analysis.
Extracted-ion chromatograms of four tryptic peptide ions extracted from LC-MS chromatograms after BAP treatment (+BAP) or no BAP treatment (No BAP). The sequences (possible phosphorylated residues, underlined) and m/z values are indicated. The peak height was normalized against the unphosphorylated ion in +BAP or the phosphorylated ion in No BAP. The dotted line indicates the stoichiometry of the phosphorylation level determined from the BAP treatment. (a) Peptide mass and mass windows: [TFCGTPEYLAPEVLEDNDYGR]3+; 816.03 Da ± 10 ppm and [pTFCGpTPEYLAPEVLEDNDYGR]3+; 869.33 Da ± 10 ppm. (b) Peptide mass and mass windows: [RPHFPQFSYSASSTA]2+; 841.90 Da ± 10 ppm and [RPHFPQFpSYSASSTA]2+; 881.88 Da ± 10 ppm.

Identification of PKBα phosphorylation sites at Thr308, Thr312 and Ser473 by MS/MS analysis.
The tryptic peptides derived from PKBα were analyzed by MS/MS. Amino acid sequences are indicated with the detected b- and y-type series ions. The series ions are indicated by arrowheads and m/z. Ions with neutral loss phosphate (-98 Da) are indicated by asterisks. (a) MS/MS spectrum of the doubly charged precursor ion at m/z 1303.49 with a Mascot expect score of 0.000353. The spectrum corresponded to the tryptic peptide 308–328 of PKBα carrying two phosphorylation moieties. The two possible phosphorylation sites were Thr308 and Thr312. (b) MS/MS spectrum of the doubly charged precursor ion at m/z 841.90. The spectrum corresponded to PKBα peptide 466–480 with a G478S substitution carrying one phosphorylation site at Ser473.
In addition to these phosphothreonines, we investigated phosphorylation at Ser473, which is required for the full activity of PKB8. We found non- and mono-phosphorylated peptides derived from the most C-terminal sequence of PKBα (RPHFPQFSYSASSTA, residues 466–480 with G478S modification) in an LC-MS analysis (Fig. 4b). The MS/MS spectrum of the mono-phosphorylated peptide showed that the N-terminal residues RPHFPQFpSYSA could be assigned by the presence of b-series ions (b2–b4, b6–b9 and b11) with phosphate specific neutral loss (-98 Da) between b8–b11, indicating that Ser473 was phosphorylated.
We also examined stoichiometry of phosphorylation using bacterial alkaline phosphatase (BAP) treatment. Comparison of the heights of the peaks from an LC-MS analysis of BAP treated and untreated specimens indicated that more than 90% of both Thr308 and Thr312 were phosphorylated (Fig. 4a). Meanwhile, a population analysis showed an approximately 30% phosphorylation rate at Ser473 (Fig. 4b). We thus concluded that phosphorylation on Thr308 and Ser473, a prerequisite for activation, were accomplished in silkworms using co-expression system.
Discussion
The BmNPV bacmid-silkworm expression system enables production of recombinant PKBα from larval fat bodies only 5 ~ 6 days after injection of the bacmid to the fifth-instar larvae. We were able to obtain approximately 1 mg of purified PKBα protein from 20 larvae. Literatures reported that the yields of GST-PKBα (kinase domain) and GST-PKBβ (kinase domain) were 32 mg and 5–15 mg/Liter culture, respectively32,33. Of note, these values are reported as GST-fusion proteins. To obtain pure cleaved PKBα, more steps are needed (cleavage and further purification) and thus the yield of pure cleaved PKBα from Sf9 is expected to be several mg/Liter culture deduced from these reports. Since 20 larvae provide 1 mg pure cleaved PKBα, we can estimate that a breeding 100 larvae is almost equivalent to 1 L culture of insect cell. To breed 100 larvae, one desktop box incubator is enough and the handling of larvae is very easy. It should be noticed that fifth-instar larvae are commercially available and we do not need to care for eggs. Thus, BmNPV bacmid-silkworm system is a really time-saving method, as compared to the cell culture. Notably, we do not need make higher titer virus and we have only to prepare bacmid using E. coli BmDH10bac. Furthermore, our analyses indicated that the recombinant protein maintained structural integrity. Thus, the BmNPV bacmid-silkworm is a viable approach for PKB production. By contrast, our attempts to obtain PKB in the soluble fraction using the E. coli expression system failed due to the formation of an inclusion body (data not shown). In the BmNPV bacmid-silkworm expression system, active PKB was successfully produced after co-expression of PDK1. Our phosphorylation assay using GSK3α/β peptide as the substrate showed that the PKBα produced in this system was slightly more active than that available commercially.
An important aspect of the system is the low cost and its relative simplicity25,26,27,28,29,30,31,34,35. The cost of feeding the silkworms is very low, with a total cost for 20 larvae of about 20 US dollars. Thus, it costs little over 20 US dollars to obtain about 1 mg of active PKBα. The cost of this approach is comparable to that of an expression system using E. coli.
Our MS analysis showed a phosphorylation rate of more than 90% at Thr308 on the activation loop of the recombinant PKB produced by the BmNPV bacmid-silkworm system (Fig. 4a). Phosphorylation of Ser473 is also thought to be essential for full activation of PKB. Here, we found a rate of Ser473 phosphorylation of approximately 30%, which may have decreased the activity of the PKBα. Nevertheless, our phosphorylation assay showed higher activity in the recombinant PKBα produced by our system than in commercially available PKBα. Although the solution to this enigma is not clear, one possible explanation is that the additional phosphorylation on the activation loop may have contributed to this higher activity. Our MS analysis showed over 90% phosphorylation of Thr312 in addition to Thr308. The importance of phosphorylation of Thr312 for activation is not yet clear; however, this phosphorylation might have changed the conformation of the activation loop and contributed to the relatively high activity of our recombinant PKBα. In the crystal structure, Thr312 is very closely located to the substrate GSK-3 peptide (Fig. S1). Further research into this aspect of PKB phosphorylation is required. It is notable that Thr312 is known to be O-GlcNAcylated36, implying that Thr312 is accessible to modifiers. In addition, Chen et al reported that Tyr315, which is also located on the activation loop, was phosphorylated and that this Tyr-phosphorylation elevated PKB activity in mammalian cells37. We did not identify this Tyr phosphorylation in the recombinant PKBα produced by our BmNPV bacmid-silkworm expression system. This discrepancy might be due to differences between the types of cell (species) used in the experiments.
Our data indicates that the PKBα recovered from silkworm is phosphorylated at its most important residue, Thr308 and has potential to be used in structural and biochemical studies. However, the recovered PKBα should currently be used with some caution in research because of the uncertainty over inhomogeneity, such as the 30% phosphorylation of the Ser473 residue. In mammalian, this phosphorylation is thought to be introduced by kinases such as Target of rapamycin complex 2 (TORC2), DNA-PK or ILK12. The relatively high rate of the phosphorylation without co-expression of Ser473 kinases could be explained by some specific physiological status in the fat body cells of growing silkworm larvae, for instance, activated TOR signaling. It should be noticed that Bombyx mori genes for the orthologous proteins for mTOR and ILK are found (BGIBMGA008952 and BGIBMGA012119, respectively) in genome database of the silkworm (http://silkworm.swu.edu.cn/silkdb/). Thus, 30% phosphorylation on S473 could be explained by endogenous Bombyx orthologs of mTOR and/or ILK. Although none of detailed research for TOR signaling in Bombyx larvae was reported, it was known that TOR signaling is activated in the fat body of Drosophila larva38. Thus, TOR pathway in the fat body cells of the silkworm is probably also activated. Unfortunately, inhomogeneity of phosphorylation may hamper crystallization, or introduce errors into interpretations of functional mechanisms in pharmaceutical assays. The inhomogeneity could be overcome using further separation on high resolution ion-exchange chromatography columns. Possibly, site-direct mutagenesis in which Thr312 and/or Ser473 residues to Glu/Asp (phosphorylation mimic) or Ala may be a promising means of diminishing the inhomogeneous phosphorylation. In future studies, co-expression of Ser473 kinase, such as Target of rapamycin complex 2 (TORC2) and/or ILK, may increase the rate of phosphorylation at Ser473.
In conclusion, this simple and rapid BmNPV bacmid-silkworm production system will be highly valuable for future high throughput drug screening as well as for biochemical and structural studies of PKBα.
Methods
Construction of the BmNPV bacmids co-expressing PKBα and PDK1 and expressing only PKBα
The donor vector pFastGSTPKB-PDK1 was constructed to co-express PKBα and PDK1. The cDNAs of the kinase domain (residues 145–481) of human PKBα and the kinase domain (residues 73–358) of human PDK1 were amplified from HeLa QUICK-Clone™ cDNA (Clontech) using the PrimestarMax DNA polymerase (Takara). PCR fragments encoding the kinase domain of PKBα fused to glutathione S transferase (GST) and the kinase domain of PDK1 were respectively cloned into regions controlled by the polyhedrin and p10 promoters of the pFastBac™ Dual vector (Invitrogen) (Fig. 1b). We also constructed the donor vector pFastGSTPKB by cloning a PCR fragment encoding the kinase domain of PKBα, fused to GST at its N-terminal, into the pFastBac™ vector (Invitrogen) (Fig. 1b). E. coli BmDH10Bac competent cells25 were transformed by pFastGSTPKB or pFastGSTPKB-PDK1 and the transformants were cultured. The BmNPV bacmids were purified using a midi prep kit (QIAGEN). The integrity of the plasmid sequence was verified by DNA sequencing.
Expression of the PKBα kinase domain in silkworm larvae and protein purification
Each BmNPV bacmid (1 µg) was suspended in 3 μl of DMRIE-C reagent. The mixture was diluted to 50 μl with water and was then injected into the dorsal side of the silkworm larvae at the first day of the fifth instar larval stage. The larvae were allowed to develop for 5 or 6 days, then their fat bodies were collected and immediately placed in PBS buffer containing 0.5% sodium thiosulfate. The washed fat bodies were then transferred to buffer A (20 mM Tris-HCl (pH 8.0), 100 mM NaCl) and lysed by sonication. The lysate was centrifuged and the supernatant was loaded onto a glutathione sepharose 4B column (GE Healthcare) and eluted with buffer containing 30 mM reduced glutathione, 50 mM Tris-HCl (pH 7.5), 400 mM KCl, 0.1 mM EDTA and 1 mM dithiothreitol (DTT). The N-terminal GST-tag was cleaved by HRV-3C proteinase. The target protein was purified by size exclusion chromatography (HiLoad 26/60 Superdex 75 pg, GE Healthcare) using a buffer containing 50 mM potassium phosphates (pH 7.5) and 20 mM KCl, 1 mM DTT and 0.1 mM EDTA. Residual GST dimer was removed by re-passing through a glutathione sepharose 4B column (GE Healthcare). The identity and integrity of the final protein sample were confirmed by SDS-PAGE. Fractions containing PKB were concentrated using an Amicon Ultra-15 (10,000 Da MWCO) concentrator (Millipore).
NMR spectroscopy
The phosphorylated PKBα was dissolved in 50 mM potassium phosphate buffer (pH 7.0) containing 100 mM KCl, 0.1 mM EDTA and 1 mM TCEP in 93% H2O/7% 2H2O. The final protein concentration was 50 μM. A 1H NMR spectrum was acquired at 25°C using an AVANCE III 600 NMR spectrometer equipped with a TCI cryogenic probe (Bruker). 1H 1D NMR was performed with water suppression by WATERGATE and water flip back techniques. The time domain data were processed using NMRPipe39.
Protein digestion and phosphatase treatment
Three hundred μg of purified PKBα was S-carbamoylmethylated using 2-iodoacetoamide (Wako)40 and desalted by methanol-chloroform precipitation41. The desalted protein was digested overnight with 3 μg of trypsin (Promega) at 37°C. One μg of the carbamoylmethylated protein was treated with 0.6 units of bacterial alkaline phosphatase (BAP, Toyobo) or same volume of water. After 2 hours incubation at 37°C, the reaction was stopped by adding 1/100 volume of formic acid.
MS analysis of the PKBα
Peptide samples were analyzed on a direct nanoflow liquid chromatography (LC) system equipped with a hybrid quadrupole-orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific) as described previously42. MS/MS data was processed with the MASCOT algorithm (version 2.2.1., Matrix Science Ltd., London, United Kingdom) to assign peptides using the UniProt sequence database (release 2011, limited to human taxonomy) and the following parameters. The fixed modification was carbamoymethyl (Cys) and the variable modification parameters were phosphorylation (Ser, Thr and Tyr), pyroglutamylation (Gln), acetylation (protein N-terminus) and oxidation (Met). The maximum missed cleavage was set at 3 with a peptide mass tolerance of +/− 15 ppm. Peptide charges from +2 to +4 states and MS/MS tolerances of +/− 0.8 Da were allowed. The MS/MS signal assignments were confirmed manually. All results of peptide searches were extracted using STEM software43. Determination of absolute phosphorylation stoichiometry was calculated by comparing the peak height of MS spectra of the native unphosphorylated peptide and the BAP treated dephosphorylated peptide. MS data including peak heights was processed by Xcalibur (Thermo Fisher Scientific). Prior to the analyses, the quantitative dynamic range of the LC-MS system was confirmed to be linear between 1.0 × 106 and 3.0 × 109 for signal height of the mass spectrum by measuring a standard peptide mixture (data not shown).
Kinase assay of PKBα
To estimate kinase activity of the PKBα, we used an Akt kinase assay kit (Cell Signaling Technology) that detects phosphorylation with nonradioactive reagents. The GSK-3 fusion protein was phosphorylated as a substrate for PKBα in the assay. The amount of phosphorylation of GSK-3 was measured by Western blotting in which the phosphorylation was recognized by a phospho-GSK-3α/β (Ser21/9) antibody. After the primary antibody was bound to the target protein, a secondary antibody, HRP-linked anti-rabbit IgG (Dako) was applied. Bound HRP activity was monitored by chemiluminescence using ImmunoStar LD (Wako).
References
Manning, B. D. & Cantley, L. C. AKT/PKB signaling: navigating downstream. Cell 129, 1261–74 (2007).
Testa, J. R. & Tsichlis, P. N. AKT signaling in normal and malignant cells. Oncogene 24, 7391–3 (2005).
Brugge, J., Hung, M. C. & Mills, G. B. A new mutational AKTivation in the PI3K pathway. Cancer Cell 12, 104–7 (2007).
Tokunaga, E. et al. Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets 8, 27–36 (2008).
Cicenas, J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers 23, 1–9 (2008).
Brazil, D. P. & Hemmings, B. A. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 26, 657–64 (2001).
Scheid, M. P. & Woodgett, J. R. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol 2, 760–8 (2001).
Alessi, D. R. et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15, 6541–51 (1996).
Alessi, D. R. et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 7, 776–89 (1997).
Alessi, D. R. et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7, 261–9 (1997).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–101 (2005).
Bayascas, J. R. & Alessi, D. R. Regulation of Akt/PKB Ser473 Phosphorylation. Molecular Cell 18, 143–145 (2005).
Lin, K. et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci Signal 5, ra37 (2012).
Lippa, B. et al. Synthesis and structure based optimization of novel Akt inhibitors. Bioorg Med Chem Lett 18, 3359–63 (2008).
Wu, W. I. et al. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One 5, e12913 (2010).
Yang, J. et al. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell 9, 1227–40 (2002).
Yang, J. et al. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol 9, 940–4 (2002).
Addie, M. et al. Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin -4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J Med Chem 56, 2059–73 (2013).
Ashwell, M. A. et al. Discovery and optimization of a series of 3-(3-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: orally bioavailable, selective and potent ATP-independent Akt inhibitors. J Med Chem 55, 5291–310 (2012).
Bencsik, J. R. et al. Discovery of dihydrothieno- and dihydrofuropyrimidines as potent pan Akt inhibitors. Bioorg Med Chem Lett 20, 7037–41 (2010).
Blake, J. F. et al. Discovery of pyrrolopyrimidine inhibitors of Akt. Bioorg Med Chem Lett 20, 5607–12 (2010).
Freeman-Cook, K. D. et al. Design of selective, ATP-competitive inhibitors of Akt. J Med Chem 53, 4615–22 (2010).
Kallan, N. C. et al. Discovery and SAR of spirochromane Akt inhibitors. Bioorg Med Chem Lett 21, 2410–4 (2011).
Xu, R. et al. Discovery of spirocyclic sulfonamides as potent Akt inhibitors with exquisite selectivity against PKA. Bioorg Med Chem Lett 21, 2335–40 (2011).
Motohashi, T., Shimojima, T., Fukagawa, T., Maenaka, K. & Park, E. Y. Efficient large-scale protein production of larvae and pupae of silkworm by Bombyx mori nuclear polyhedrosis virus bacmid system. Biochem Biophys Res Commun 326, 564–9 (2005).
Na, Z. et al. Efficient production of canine interferon-alpha in silkworm Bombyx mori by use of a BmNPV/Bac-to-Bac expression system. Appl Microbiol Biotechnol 78, 221–6 (2008).
Sakamoto, S. et al. Efficient silkworm expression of single-chain variable fragment antibody against ginsenoside Re using Bombyx mori nucleopolyhedrovirus bacmid DNA system and its application in enzyme-linked immunosorbent assay for quality control of total ginsenosides. J Biochem 148, 335–40 (2010).
Sasaki, K. et al. Silkworm expression and sugar profiling of human immune cell surface receptor, KIR2DL1. Biochem Biophys Res Commun 387, 575–80 (2009).
Kato, T., Kajikawa, M., Maenaka, K. & Park, E. Y. Silkworm expression system as a platform technology in life science. Appl Microbiol Biotechnol 85, 459–70 (2010).
Kajikawa, M. et al. Efficient silkworm expression of human GPCR (nociceptin receptor) by a Bombyx mori bacmid DNA system. Biochem Biophys Res Commun 385, 375–9 (2009).
Kato, T., Thompson, J. R. & Park, E. Y. Construction of new ligation-independent cloning vectors for the expression and purification of recombinant proteins in silkworms using BmNPV bacmid system. PLoS One 8, e64007 (2013).
Fabbro, D. et al. Homogeneous purification of human recombinant GST-Akt/PKB from Sf9 cells. Protein Expr Purif 17, 83–8 (1999).
Baer, K. et al. Activation of a GST-tagged AKT2/PKBbeta. Biochim Biophys Acta 1725, 340–7 (2005).
Dong, J. et al. Expression and purification of bioactive hemagglutinin protein of highly pathogenic avian influenza A (H5N1) in silkworm larvae. J Virol Methods 194, 271–6 (2013).
Honjo, E. et al. Expression of the extracellular region of the human interleukin-4 receptor alpha chain and interleukin-13 receptor alpha1 chain by a silkworm-baculovirus system. Protein Expr Purif 60, 25–30 (2008).
Wang, S. et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS One 7, e37427 (2012).
Chen, R. et al. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem 276, 31858–62 (2001).
Grewal, S. S. Controlling animal growth and body size - does fruit fly physiology point the way? F1000 Biol Rep 4, 12 (2012).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–93 (1995).
Taoka, M. et al. Only a small subset of the horizontally transferred chromosomal genes in Escherichia coli are translated into proteins. Mol Cell Proteomics 3, 780–7 (2004).
Wessel, D. & Flugge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138, 141–3 (1984).
Taoka, M. et al. An analytical platform for mass spectrometry-based identification and chemical analysis of RNA in ribonucleoprotein complexes. Nucleic Acids Res 37, e140 (2009).
Shinkawa, T. et al. STEM: a software tool for large-scale proteomic data analyses. J Proteome Res 4, 1826–31 (2005).
Acknowledgements
This work was supported by KAKENHI: Grant-in-Aid for Scientific Research on Innovative Areas, Structural Cell Biology, Transient Macromolecular Complex and Protein Modifications in Pathogenic Dysregulation of Signaling, from the MEXT. R.M. is a recipient of Grant-in-Aid for JSPS Fellows.
Author information
Authors and Affiliations
Contributions
R.M. and T.K. constructed the BmNPV bacmid-silkworm system and performed the PKB expression and purification under instruction from K.M. and M.M. R.S. performed the phosphorylation assay. M.T. and C.F. performed the MS analysis. R.M., T.F., Y.I., T.I., T.H. and M.M. designed the research. R.M., T.A. and M.M. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY INFO
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Maesaki, R., Satoh, R., Taoka, M. et al. Efficient and cost effective production of active-form human PKB using silkworm larvae. Sci Rep 4, 6016 (2014). https://doi.org/10.1038/srep06016
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06016
This article is cited by
-
Humoral immune response induced with dengue virus-like particles serotypes 1 and 4 produced in silkworm
AMB Express (2022)
-
Characterization of Recombinant Thermococcus kodakaraensis (KOD) DNA Polymerases Produced Using Silkworm-Baculovirus Expression Vector System
Molecular Biotechnology (2017)
-
Efficient expression of single chain variable fragment antibody against paclitaxel using the Bombyx mori nucleopolyhedrovirus bacmid DNA system and its characterizations
Journal of Natural Medicines (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
