
Abstract
Quantification of basic cell functions is a preliminary step to understand complex cellular mechanisms, for e.g., to test compatibility of biomaterials, to assess the effectiveness of drugs and siRNAs and to control cell behavior. However, commonly used quantification methods are label-dependent and end-point assays. As an alternative, using our lensfree video microscopy platform to perform high-throughput real-time monitoring of cell culture, we introduce specifically devised metrics that are capable of non-invasive quantification of cell functions such as cell-substrate adhesion, cell spreading, cell division, cell division orientation and cell death. Unlike existing methods, our platform and associated metrics embrace entire population of thousands of cells whilst monitoring the fate of every single cell within the population. This results in a high content description of cell functions that typically contains 25,000 – 900,000 measurements per experiment depending on cell density and period of observation. As proof of concept, we monitored cell-substrate adhesion and spreading kinetics of human Mesenchymal Stem Cells (hMSCs) and primary human fibroblasts, we determined the cell division orientation of hMSCs and we observed the effect of transfection of siCellDeath (siRNA known to induce cell death) on hMSCs and human Osteo Sarcoma (U2OS) Cells.
Similar content being viewed by others
Introduction
Though microscopy is gaining deeper access inside the cell, appropriate methodologies for cell monitoring at a mesoscopic scale with strong statistics both in space and time are still missing. Real-time cell culture monitoring is essential in cases where the behavior of not just a single cell but a cell population dynamics needs to be observed with significant temporal resolution. Various imaging platforms have been explored to meet this requirement, especially, video microscopy and impedance readers1,2,3,4,5,6. Limited field of view, high cost and complexity in manipulating cell culture during the experiment, are the major limitations of video microscopy. Further, in most of the cases, labeling is required for visualization and analysis, which raises concerns regarding photo-toxicity and experimental bias7. Substrate impedance measurement overcomes these limitations. However, it is an indirect approach. First, the obtained parameters are surrogate measurements of substrate impedance changes. Second, the measurement is restricted to cell population and is not usually extended to the level of single cells. Third, the cells are not visualized which represents a huge loss of information in the era of HCA. As a recent alternative, owing to its simplicity, lensfree imaging is being assessed to perform live cell imaging8,9,10,11. Using our lensfree video microscopy platform (methods, Fig. 1, Fig. 2) compatible with standard 35 mm culture dish, we reported a real-time, label-free method for the detection of dividing cells in a population of thousands of cells10.

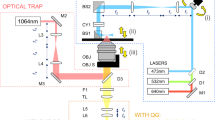
Lensfree video microscopy platform.
(a) Schematic diagram explaining the principle of lensfree imaging. (b) Lensfree video microscope consisting of LED, Pinhole, 24 mm2 CMOS imaging sensor and temperature control module. (c) Raw image obtained from the culture of hMSCs imaged by lensfree video microscope also showing a magnified region. The field of view of the entire image is 24 mm2 containing ~ 3700 cells.

Real-time cell culture monitoring inside standard incubator.
Photograph showing 4 lensfree video microscopes inside the standard incubator in parallel. The culture dishes placed on the imaging sensors have a diameter of 35 mm.
In this article, we demonstrate the capability of our lensfree video microscope to monitor the fundamental processes of the cell culture directly inside a standard incubator. We introduce specifically devised metrics to follow cell-substrate adhesion, cell spreading, cell division, cell division orientation and cell death. We show that these metrics can be applied to a very large range of population, from few tens to more than 4000 cells, for a period ranging from few hours to weeks. More notably, these metrics allow following the fate of single cells within large populations and large period of observations. Our methodology consisted in first testing and assessing different metrics at the level of single cells, followed by computation of the metrics over the entire population as a function of time. This resulted in scatter plots compiling 25,000–900,000 label-free measurements depending on cell density and period of observation. As proof of concept, we studied the major cell functions of primary human fibroblasts, human Mesenchymal Stem Cells (hMSCs) and human Osteo Sarcoma (U20S) cells.
In sum, we show that along with dedicated image processing, our lensfree video microscope offers a robust platform to quantitatively and non-invasively follow 2D cell culture in real-time, with high statistical significance. The setup and associated methods are thus suitable for myriad applications including high-throughput screening, biocompatibility assays, etc.
Results
Cell-substrate adhesion and spreading
The process of cell-substrate adhesion captured by means of lensfree video microscope is seen in Fig. 3a. Lensfree hologram of a floating cell is similar to airy pattern (Fig. 3a at t0, t0 + 0h40m and t0 + 1h20m, Supplementary Fig. 1). The zero order is darker, with relatively lower gray values: less than 100 gray levels (with the maximum value of 255 gray levels and background of ~70 gray levels) and several interference rings are observed. As the cell adheres to the substrate, there is a sharp change in the corresponding lensfree hologram (Fig 3a at t0 + 2h, Supplementary Fig. 1). In contrast to the lensfree hologram obtained from a floating cell, here the zero order gray value increases 2 to 3 folds, reaching larger values: typically values greater than 200 gray levels. Supplementary Fig. 1 shows that the difference between floating and adhered cells is profound in images obtained from lensfree video microscope compared to lens-based microscope.

Cell-substrate adhesion.
(a) Time lapse images showing cell substrate adhesion of a single primary human fibroblast. The images at t0, t0 + 0h40m, t0 + 1h20m shows the floating cell. At t = t0 + 2h, the cell adhered to the substrate shown by the increase in the zero order gray value. Scale bar 50 μm. (b), (c) Cell substrate adhesion kinetics of hMSCs and primary human fibroblasts respectively (N = 3 experiments). In the case of hMSCs, 90% of population adhered to the substrate in 5h30m ± 30m (S.D., N = 3 experiments). Whereas, cell substrate adhesion of primary human fibroblasts was slower and 90% adhesion was achieved after 8h40m ± 40m (S.D., N = 3 experiments). Region A denotes rapid adhesion immediately after plating. Region B denotes the decrease in the adhesion rate after ~60% of the population adhered to the substrate, while region C shows the plateau after nearly 95% of the cells adhered to the substrate.
Therefore, by using gray value differentiation and pattern recognition, floating cells and adherent cells can be detected, counted and quantified (methods, Supplementary video 1). The percentage of floating and adherent cells can be obtained at each time point thus providing the kinetics of cell adhesion. As proof of concept, we monitored cell-substrate adhesion kinetics of hMSCs and primary human fibroblasts (Fig. 3b, 3c). In the case of hMSCs, 90% of population adhered to the substrate in 5h30m ± 30m (S.D., N = 3 experiments). Whereas, cell substrate adhesion of primary human fibroblasts was slower and adhesion of 90% of the population was achieved in 8h40m ± 40m (S.D., N = 3 experiments). We noted that the adhesion kinetics of both hMSCs and primary human fibroblasts followed the same trend. That is, immediately after plating, the percentage of cell adhesion increased rapidly (Fig. 3b, 3c, Region A) until reaching a certain value (adhesion of ~60% of the population) after which the adhesion rate became slower (Fig. 3b, 3c, Region B). After ~95% of cell substrate adhesion, a plateau was reached (Fig. 3b, 3c, Region C).
Following this ‘passive event’ of initial adhesion with the substrate, the cells proceeded to the ‘active event’ of spreading. By using appropriate metrics to extract information from both raw (Fig. 4a) and reconstructed (Fig. 4b) images, in addition to merely distinguishing floating and adherent cells, we monitored the kinetics of the entire process of cell adhesion and spreading. The metrics that we propose are kurtosis from the raw image and aspect-ratio obtained after holographic reconstruction of the raw lensfree holograms.

Cell-substrate adhesion and spreading.
(a) Time lapse images of raw lensfree holograms of adhering primary human fibroblast cell. Scale bar 50 μm. (b) Lensfree holographic reconstruction and segmentation of (a). Scale bar 50 μm. Note that at t0, since the cell was afloat, the distance between the cell and the imaging sensor was larger compared to the subsequent frames. However, this difference was not considered while performing holographic reconstruction. (c) Kurtosis measurement of 10 cells along with the mean (blue). Rise time of the mean corresponds to the average time taken for the cells to adhere to the substrate (region A) and the decay time corresponds to the average time taken for the cells to spread (region B). Region C denotes stable kurtosis values post cell spreading. (d) Aspect-ratio measurement of 10 cells along with the mean (blue). The absence of values at t = 0h00m and t = 0h20m denotes that the cells were afloat. The decay time corresponds to the average time taken for cell spreading (Region B). (e),(g) Scatter plots containing ~100,000 kurtosis values of nearly n = 5000 primary human fibroblasts and hMSCs from N = 3 experiments, respectively. The mean is plotted in white dotted line. Region A denotes the initial adhesion of cells to the substrate, region B denotes time taken for the cells to spread and region C shows the stability of the kurtosis values post cell spreading. Error margin in regions A, B and C, is the standard deviation resulting from N = 3 experiments. (f),(h) Scatter plots of ~100,000 aspect-ratio values obtained from primary human fibroblasts and hMSCs over N = 3 experiments respectively, along with the mean (in white dotted line). Region B shows the time taken for cell spreading. Error margin in regions A, B and C, is the standard deviation resulting from N = 3 experiments.
Though during the initial cell-substrate contact, the intensity change in the raw image is predominant (Fig. 4a, t = t0 + 1h00m), the change becomes subtle during the process of cell spreading (Fig. 4a t0 + 1h30m until t0 + 4h). However, variation is measured by computing kurtosis of the lensfree hologram obtained from the cells (methods). Kurtosis is a way to determine whether the gray value distribution of an image is almost peaked or flat. First, we plotted the changes in kurtosis, with respect to time, obtained from 10 adhering primary human fibroblasts. From figure 4c, it can be observed that, typically the kurtosis of a floating cell is between 4 a.u. and 8 a.u. describing a rather flat gray value distribution. The distribution is peaked, due to the sharp change in the zero order gray value during the initial attachment of the cell to the substrate: kurtosis increases 3 to 6 folds up to 40 a.u. As the cell spreads, the zero order gray value decreases resulting in a flat distribution. Kurtosis descends gradually attaining a value of 10.4 ± 2.7 a.u (S.D., n = 10 cells) and remains stable.
By determining the rise time and decay time of mean kurtosis (Fig. 4c, blue), we systematically characterized the kinetics of cell adhesion and spreading. Rise time is defined as the time taken for a response to rise from 10% to 90% of the final value. In other words, rise time corresponds to the increase in kurtosis resulting from cell substrate adhesion (Figure 4c, region A). Decay time is defined as the time taken for a response to fall from 90% to 10% of the final value. Therefore, decay time corresponds to the time taken for the cells to spread (Figure 4c, region B). Here, the rise time was 40 minutes (Fig. 4c, region A, t = 0h00m to t = 0h40m) and the decay time of 6 hours commenced at t = 2h20m and lasted until t = 8h20m (Fig. 4c, region B). This shows that, on an average (n = 10 cells), the time taken for the cells to adhere to the surface was 40 minutes and to spread was 6 hours.
We extended this measurement to the entire population and compared the cell spreading time of hMSCs and primary human fibroblasts. This resulted in scatter plots (Fig. 4e, 4g), in each case containing more than 100,000 measurements obtained from nearly 5,000 cells (N = 3 experiments) followed over a period of 20 hours. We calculated the rise time and decay time for the mean curves of the scatter plots (white dotted line, Fig. 4e, 4g), to obtain initial cell adhesion time and cell spreading time respectively.
Initial adhesion time was similar for both hMSCs and primary human fibroblasts with values of 1h00m ± 20m (S.D., N = 3 experiments) and 1h00m ± 30m (S.D., N = 3 experiments) respectively (Fig. 4e, 4g: region A). For hMSCs, cell spreading started at t = 2h00m ± 30m (sd, N = 3 experiments) and lasted until 11h00m ± 30m (S.D., N = 3 experiments) (Fig. 4e, 4g: region B). For primary human fibroblasts, cell spreading started at t = 1h20m ± 20m (S.D., N = 3 experiments) and lasted until 8h20m ± 40m (S.D., N = 3 experiments).
The second method consists in calculating the aspect-ratio of the cells from the reconstructed image (Fig. 4b, Supplementary video 2). Aspect-ratio is the ratio of the minor axis length to the major axis length of the ellipse which best fits the shape of the cell segmented from the reconstructed image (methods). The aspect-ratio takes a value between 0.0 a.u. and 1.0 a.u, with 1.0 being perfectly circular and < 0.1 being highly elongated. When a cell first contacts the surface, it is circular in shape with an aspect-ratio between 0.9 a.u and 1.0 a.u. As the cell spreads to adopt a more elliptical shape, the aspect-ratio decreases.
The time dependent change in the aspect-ratio of single spreading cells (n = 10 single cells) is shown in Fig. 4d. The aspect-ratio was calculated only after the cells adhered to the substrate. Consequently, the absence of the initial values (Fig. 4d, t = 0h00m and t = 0h20m) indicates that the cells were afloat. The decay time of the mean (Fig. 4d, blue) corresponds to average cell spreading time of 10 single cells. The decay time obtained is 6h20m starting from t = 1h00m, until t = 7h20m.
Further, we measured the aspect-ratio for the entire population of hMSCs and primary human fibroblasts (Fig. 4f, 4h). The aspect-ratio measurement was performed after nearly 50% cells adhered to the substrate (t = 0h40m for primary human fibroblasts and t = 0h30m for hMSCs). Due to substantial diversity of the population (Supplementary Fig. 2), the mean aspect-ratio only reached a maximum of ~0.8 a.u. even during the initial stages of adhesion. Although, from the scatter plots (Fig. 4f, 4h) it can be observed that more than 50% of the population had an aspect-ratio value greater than 0.8 a.u. at the beginning. HMSCs started to spread at 1h30m ± 30m (S.D., N = 3 experiments) and were almost completely spread by 13h00m ± 1h (S.D., N = 3 experiments) denoted by a stable aspect-ratio value of 0.3–0.2 a.u. (Fig. 4f: Region B). In the case of primary human fibroblasts, cell spreading started at t = 1h40m ± 20m (S.D., N = 3 experiments) and lasted until 10h40m ± 1h (S.D., N = 3 experiments) (Fig. 4h: Region B).
Cell division
First the cells adhere to the substrate and spread well, next, they move11 and proliferate (Supplementary Video 3). Figure 5a and 5b shows the lensfree holograms obtained from a dividing hMSC and its reconstruction respectively. The morphological changes undergone by the cell are observed from t = t0 until t0 + 1h00m. Longest axis prior to cell rounding is denoted by dotted yellow line at t0 + 0h40m. Round cell with reduced cell-substrate adhesion is observed at t0 + 1h20m. The axis of cell division (Fig. 5a, 5b: red dotted line) and the 2 daughter cells are observed at t = t0 + 1h40m and t0 + 2h00m respectively.

Cell-division orientation.
(a) Time-lapse raw lensfree holograms obtained from a dividing hMSC obtained over 02h20m. Yellow dotted line at t = t0 + 0h40m denotes the longest axis prior to cell rounding. The green circle at t0 + 1h20m indicates the rounded, partially detached metaphase cell. The red dotted line at t = t0 + 1h40m shows the cell division axis. Scale bar 50 μm. (b) Holographic reconstruction and segmentation of (a). Note that the change in the distance between the cell and the imaging sensor during mitotic cell rounding (t = t0 + 1h20m) was not taken in to account while performing holographic reconstruction. This resulted in artifacts around the cell observed in the holographic reconstruction at t = t0 + 1h00m. Scale bar 50 μm. (c) Rose plot showing the difference in angles between the longest axis and the cell division axis calculated from 299 cell divisions over a period of 4 days.
We have previously demonstrated that all dividing cells exhibit a particular holographic pattern corresponding to mitotic cell rounding (Fig. 5a, t0 + 1h20m) and that dividing cells in a population can be identified and numbered in real-time by detecting these specific patterns10. The detection of patterns to identify dividing cells is performed on the raw image. We noted that reconstructing the images obtained from a dividing cell (Fig. 5b), did not enhance the detection of the dividing cell. Again pattern recognition had to be employed and the signal was not enhanced in any way. However, holographic reconstruction permitted further examination of cell division process. i.e., it allowed capturing the orientation of cell division.
As a proof of principle, we determined the difference in angles between the longest axis and the axis of cell division of dividing hMSCs over a period of 4 days (Fig. 5c, methods). The longest axis of the cell was determined from the reconstructed images obtained prior to mitotic cell rounding (Fig. 5b, t0 + 0h40m: longest axis denoted by dotted yellow line) and the cell division axis was determined from the image following cell-rounding (Fig. 5b at t = t0 + 1h40m: cell division axis denoted by red dotted line). In total, we analyzed 299 cell divisions from 208 images obtained over a period of 3 days. From the rose plot (or angle histogram chart) (Fig. 5c), it is seen that, ~46% of the cells divided within a 30° wide sector with respect to their longest axis. In ~35% of cell divisions, the difference between the longest axis and the axis of cell division was between 30° and 60°. Owing to the large statistics, we are also able to distinguish a relatively small percentage of the population (~19%) that exhibited a difference of more than 60° between longest axis and the axis of cell division.
Cell death
We introduce 2 metrics for the detection of cell death using lensfree video microscope. The first metric monitors cell death by detecting the changes in the gray value associated with cell detachment. The method is applied on raw image and does not require holographic reconstruction. The second method employs holographic reconstruction to detect the changes in cell morphology to monitor cell death. We followed cell death kinetics on 2 different cell types: U2OS and hMSC. In both the cases, cell death was induced by forward transfection using the positive cell death phenotype control siRNA (siCellDeath) (Methods).
Single U2OS cell experiencing cell death, upon introduction of siCellDeath, is seen in Fig. 6a. At t = t0 + 35h, the gray value of the cell changes instantaneously as it detaches from the substrate. As stated earlier, the gray value at the center of the hologram (zero order gray value) obtained from a floating cell is much lower compared to that of an adherent cell. Thus change in zero order gray value is expected when cells detach during cell death (Supplementary video 4, Supplementary Fig. 3). Figure 6b shows the change in the gray value of 10 U2OS cells during cell death. The transfection was performed at t = t0. After 28 hours, the gray values plummeted from 210.7 ± 39.6 gray levels (S.D.) to 70.4 ± 20.3 gray levels (S.D.) emphasizing the detachment of the cells from the substrate.

Cell death – human Osteo Sarcoma (U2OS) cells.
(a) Time-lapse lensfree holograms obtained from a dying U2OS cell. The cell detaches from the substrate at t = t0 + 35h (t0 = immediately after transfection), visible from the change in the gray value. Scale bar 50µm. (b) Change in gray value associated with cell death of 10 U2OS cells along with its mean. siRNA transfection was performed at t = 0h. (c) Scatter plot containing >900,000 gray values obtained from 3506 ± 228 cells over a period of 90 hours. Red arrow denotes the moment of siRNA transfection. Region B denotes the time taken for more than 80% of the cell population to die. Region C shows that the detached cells did not reattach to the substrate. The error margin in regions B, C, is the standard deviation resulting from the local fluctuations in the cell population (n ~ 3500 cells).
It is further illustrated in a much larger scale from the scatter plot of the entire population containing more than 900,000 measurements from 3506 ± 228 cells (S.D. resulting from local variation in cell counting over the period of observation) over a period of 90 hours (Fig.6c). The scatter plot represents the cell population from the time of cell plating to the time of cell death. Initially at t = t0-20h, the cells were afloat with gray values between 60 and 100 gray levels (Fig. 6c, region A). Within first 10 hours (from t = t0-20h to t = t0-10h), more than 75% of the cells adhered to the substrate, as shown by the increase in gray values. T = t0 marks the moment of transfection (Fig. 6c, red arrow mark, Methods). The effect of transfection is visible in Fig. 6c, from t = t0 + 22h. At this stage, nearly 15% of the cells experienced cell death demonstrated by the accumulation of values between 60 gray levels and 100 gray levels in the scatter plot. A predominant change is observed only from t = t0 + 30h, when more than 50% of the cells experienced cell death. At t = t0 + 40h, the distribution completely reversed compared to t = t0 (immediately after transfection) as ~80% of the cells detached taking gray values between 65 gray levels and 110 gray levels. Within the duration of 18h (±4h) (S.D., n ~ 3500 cells) (Fig. 6c, region B) post transfection, starting from t0 + 22h (±1h) (SD, n ~ 3500 cells) until t0 + 40h (±3h) (S.D., n ~ 3500 cells), nearly 80% of the population suffered cell death, testifying the effectiveness of siCellDeath. It is to be noted that the distribution at the start of the experiment (t = t0-20h) and at the end of the experiment (t = t0 + 70h) are similar. This shows that the gray value of live floating cells is equal to the gray value of dead detached cells. Also, region C (Fig. 6c) shows that the detached cells did not reattach to the substrate as long as 20 hours after detachment.
We reproduced the same approach to quantify cell death induced by siCellDeath on hMSCs. Fig. 7a and 7b shows the holograms obtained from a dying hMSC and the reconstructed images, respectively. It is notable from figure 7a, that the change in the gray value associated with cell death was not as significant as it was observed in U2OS cell death (Fig. 6a). In fact, although hMSCs retracted from the substrate and became circular, they did not completely detach from the substrate upon cell death (remaining subtly attached). As a consequence, the gray value remained high throughout and did not provide a precise reflection of cell death (Supplementary Fig. 4). For this reason, we measured the change in the aspect-ratio of the cells in order to efficiently quantify cell death in the case of hMSCs (Methods). Measuring aspect-ratio of cells (from the reconstructed image) provided information on the change in the morphology of cells during cell death. Fig. 7b shows the morphological change (rounding) endured by a dying hMSC, clearly visible from the reconstructed images. At t = t0, the cell was well spread with an aspect-ratio close to 0.4 a.u. As the cell became circular due to cell death, the aspect-ratio gradually a reached value close to 0.9 a.u. at t = t0 + 60h.

Cell death – hMSCs.
(a) Time-lapse lensfree holograms obtained from a dying hMSC. Time t0 marks the moment of transfection. Scale bar 50 μm. (b) Holographic reconstruction and segmentation of (a). Note that the change in the distance between the cell and the imaging sensor that may have occurred from (t = t0 + 40h until t = t0 + 70h) was not taken in to account while performing holographic reconstruction. Scale bar 50 μm. (c) Scatter plot showing the changes in the aspect-ratio of 3699 ± 215 cells over 180 hours. Region A and region B shows the time taken for cell spreading and cell death/cell rounding respectively. Error margin is the standard deviation resulting from the local fluctuation of aspect-ratio values in the cell population (n ~ 3700 cells). (d) ‘max plot’ showing the most populated range with an interval of 0.1 a.u.
A similar effect was observed in the entire population containing 3699 ± 215 cells (S.D., resulting from local variation in cell counting over the period of observation) (Fig. 7c). The cells were monitored from cell plating until cell death for a period of 7.5 days. Due to large population of cells and significant diversity, mean (of the scatter plot) did not provide an accurate description of cell death. Hence, we segregated the aspect-ratio values from 0.0 to 0.1 a.u. in to 10 ranges. We computed the most populated range (bin corresponding to the max value of the histogram) at each frame (‘max plot’) (Fig. 7d). Initially after cell plating (t = t0-90h), more than 60% of the cell population had an aspect-ratio greater than 0.8 a.u. As the cells spread, the distribution of cell aspect-ratios changed gradually (Fig. 7c, 7d). The ‘max plot’ reached 0.4 a.u and remained stable with fluctuations of ±0.1 a.u (S.D., n ~ 3700 cells). siCellDeath transfection was performed at t = t0 (Fig. 7c, 7d, red arrow mark, Methods). The aspect-ratio values changed gradually from t = t0 + 20h. The rise time of the plot corresponds to the rounding time of dying cells i.e. to the kinetics of cell death. The rise time commenced at t = t0 + 23h (±2h) (S.D., n ~ 3700 cells) hours post transfection and reached the final value (~0.9 a.u.) at t = t0 + 49h (±3h), lasting 28 ± 3 hours (Fig. 7c, 7d, region B). At this stage, it can be observed that more than 70% of the population had an aspect-ratio of 0.8 ± 0.1 a.u (S.D., n ~ 3700 cells). Region C in Fig. 7c, 7d shows that the aspect-ratio values did not descend back denoting that the cell rounding due to cell death was irreversible.
It is noteworthy that the distribution towards the end of the experiment is similar to the distribution at the start of the experiment. This states that, in general, the average aspect-ratio is the same, 0.9 ± 0.1 a.u., for both, just adhered living cells and loosely attached dead cells. The decay time and the rise time provided the time taken for the values to descend from 0.9 a.u to 0.4 a.u during spreading and then from 0.4 a.u. back to 0.9 a.u. during cell death. The spreading time of living cells (following cell plating) was 17 ± 2 hours from t = t0-88 h to t = t0-71h (±2h) (S.D., n ~ 3700 cells) (Fig. 7c, 7d: region A). The rounding time of dying cells was 28 ± 3 h (S.D., n ~ 3700 cells) (Fig. 7c, 7d: region B). This shows that, in this experiment, cell rounding of dying cells was slower compared to cell spreading of live cells.
In addition to reduction in cell size (aspect-ratio) we also monitored loss of cell motility in the case of hMSCs. We measured the average velocity of hMSCs to be 0.40 µm/min (before transfection). Interestingly, a considerable loss in cell motility was evident from nearly 18 hours post transfection (Fig. 8). The mean velocity gradually dropped from 0.40 µm/min to 0.26 µm/min in 12 hours (t0 + 30h). To the best of our knowledge, this is the first time that the reduction in cell velocity is reported in hMSCs during siCellDeath transfection. Together, the mean velocity and aspect-ratio can be precursors for cell death, observed nearly 20 hours (for hMSCs) before a known metric usually associated to cell death such as cell detachment.

Cell motility.
Mean velocity of hMSCs (n ~ 3700 cells throughout the time period) decreases due to the effect of siCellDeath (t0 marks the moment of siRNA transfection). The mean velocity started to decrease ~ 18 h post transfection.
Discussion
Lensfree imaging is being explored extensively of late. Since 2007, the Ozcan Research Group (UCLA) has described different possible geometries12,13,14 and possible diagnostic applications15,16. In addition, they have demonstrated the possibility of coupling lensfree imaging with other imaging modalities, e.g. fluorescence and reflection measurements17,18,19,20. These studies confirm the use of lensfree imaging technique as a pragmatic response to the increasing complexity of imaging systems, primarily because of its low cost and ease of use.
However, concerning cell culture inside standard incubator, little investigation has been done so far to apply lensfree imaging technique. Although several articles demonstrate the application of lensfree technique to cell imaging12,20,21,22,23,24,25,26, none of them show the possibility of using lensfree imaging for continuous cell culture monitoring inside standard incubator for extended period of observation, in a way compatible with standard practices followed by the biologists. Kim et al. demonstrated real-time detection of cardiotoxicity using lensfree imaging inside standard incubator, by measuring the variances of beating cardiomyocytes27. However, the period of observation was very short lasting ~ 2 hours and global variation in the image was measured without extending to the level of single cells. G. Zheng et al. demonstrated ‘ePetri’, a system based on lensfree shadow imaging to monitor cell culture in real-time8. The method described in the article is closer to the standard cell culture practices since it allows extended continuous monitoring inside standard incubator. However, this system requires the user to perform a skillful preparation of the cell culture within a customized sensor featuring a PDMS well. In addition, a nutrient filled fluid was used instead of normal growth substrate, in order to improve resolution. G. Jin et al. reported a lensfree imaging device for real-time cell culture monitoring but outside the standard incubator. Ambient conditions were provided to the cells by integrating oxygen permeable PDMS wall sandwiching the cover glass, custom-built heating block and an uninterrupted flow of CO2 independent media. This would increase the complexity of necessary manipulation (change of culture media, addition of drugs, etc.) of cell culture during the experiments.
Hence, so far, the needs of cell culture have been circumvented with dedicated fluidic chambers or customized sensors. Cell culture protocols were largely modified in order to adhere to the demands of lensfree imaging while focus should be laid on the contrary in order to promote the acceptability of simple and versatile lensfree imaging as a platform for real-time cell culture monitoring.
The lensfree video microscope and the associated metrics demonstrated in the paper are entirely compatible with the standard practices of cell culture, accommodating mostly used culture dishes (petri dishes of different diameter, T-flasks, multi-well plates) eliminating entirely the need for culturing cells in between microscopic slides. With our setup, the culture dish is simply placed on the device installed permanently inside the incubators, without considering distance to sensor, without preparing sample within slides or a dedicated chamber. Hence we do not compromise with the standard practices in cell culture laboratories and the overall simplicity of lensfree microscopy.
By using our lensfree video microscopy platform along with image analysis, we demonstrated different metrics to quantitatively describe different stages of a cell's life: cell adhesion, cell spreading, cell division and cell death (Fig. 9).

Major cell functions.
Schematic diagram providing a generic view of a cell life, emphasizing the major cell functions: cell adhesion, cell spreading, cell division and cell death.
Most of the cells are adherent to the substrate in nature, with few exceptions such as hematopoietic cells. Cell - substrate adhesion and spreading are complex processes involving various molecular actors and signaling pathways. These processes are categorized in to passive and active events. Passive event is the initial contact and adhesion of a cell with the substrate followed by the active event which involves in actin polymerization and myosin contraction. The ability of a cell to attach and spread has important consequences28,29. Cell-substrate adhesion is key to wound healing, tissue differentiation, etc. Study of cell - substrate adhesion and spreading is therefore inevitable to study the fundamental processes of a cell, compatibility of biomaterials, metastasis, etc. In spite of its importance, cell adhesion assays are either laborious or expensive. Most commonly used techniques either involve manual washing accounting for low reproducibility and experimental bias or involve high-cost microscopy platforms that can monitor only a few tens of cells in a population30,31,32,33. Also, one of the major drawbacks is that majority of the assays account for only one of the two processes: either cell adhesion or cell spreading.
We overcome the limitations by using lensfree video microscopy and dedicated metrics. The metrics we established to measure cell adhesion and spreading are kurtosis and aspect-ratio. To our knowledge, label-free, real-time, high-throughput method that can quantify both cell adhesion and cell spreading and that is capable of visualizing the single cells and a large population such as this has not been reported before. Though both the metrics provided similar results, one might be more sensitive compared to the other depending on the cell type, cell density, etc. As proof of concept, we compared cell-substrate adhesion and spreading of hMSCs and primary human fibroblasts. Though there was not a significant difference, it ensures that our platform and metrics can be used in a similar way to compare several substrates, cell types, effect of drugs, etc.
The orientation of cell division controls the fate of the daughter cells and therefore has important consequences. ‘Long-axis rule’ states that cells divide along their longest-axis prior to cell division, claiming that only the shape of the cell has an effect on the orientation of cell division. However, several other factors such as environment, stress, cortical cues, etc., which directly influence the orientation of cell division have been identified34,35,36,37. Extensive research is being carried out to discover other factors that may influence the orientation of cell division. By being able to detect and follow cell divisions longitudinally, lensfree video microscopy provides an apt platform to perform a detailed study on the factors influencing the orientation of cell division. To our knowledge, such a high-throughput, label-free, measurement has not been proposed before without the utilization cell localization techniques. Here, we computed the difference between longest axis and cell division axis without introducing any constraint on the cells. However, by using micro patterned substrates37 or by integrating microfluidic platforms, cells can be exposed to mechanical, or environmental stress. The influence of these factors on the axis of cell division, frequency of cell division (number of cell divisions/period of observation) and the time taken from cell rounding to separation of daughter cells can be analyzed using lensfree video microscopy.
Premature termination or uncontrolled prolongation of a cell's life is directly responsible for degenerative disorders, auto-immune diseases and cancer. Hence, factors that play a role in causing or counteracting cell death and the various mechanisms involved in cell death are extensively studied. Cell viability assays are imperative to perform these studies. Particularly, real-time monitoring is essential to study the time-dependent kinetics of cell death. A review of cell viability assays from O. Kepp et al., discusses about various assays and technological platforms that are currently used38. Out of these, only 2 platforms allow real-time monitoring of cell death, which are video microscopy and impedance reader. Limited field of view, cost, complexity and requirement of labels are limitations of video microscopy. Inability to visualize the cells and being indirect are the limitations of substrate impedance techniques. Lensfree video microscopy offers an alternative to the above mentioned methods. To monitor cell death, we used two metrics, gray value measurement and aspect-ratio measurement to provide information on cell detachment and cell rounding respectively. The kinetics is observed from the scatter plot from which the percentage of live and dead cells at any desired point in time can be obtained. Importantly, in the case of gray value measurement, there is 3σ significance in differentiating dead cell with an average gray value of 70 gray levels and a live cell with an average gray value of 210 gray levels. In the case of U2OS cells, measurement of gray value was sufficient. Whereas, in the case of hMSCs aspect-ratio measurement was needed. We monitored the effect of siCellDeath, which is well known to induce cell death. However, several other siRNAs and drugs could be tested to determine their efficacy in killing the cells. Along with the ability to follow the kinetics of cell death, recognizing the different types of cell death (apoptosis, necrosis, autophagy) is an exciting prospect.
Ironically, in the era of ‘seeing is believing’, microscopy is rarely used to monitor the above mentioned major cell functions. This is because, to perform these assays using microscopy, a personnel intervention is required to acquire images at different time points which apart from being severely labor-intensive raises concerns about sterility, incapacity to follow the same cell at single cell level. Though using video microscopy platform, the above mentioned limitations are overcome, requirement of specialized chambers and ambient conditions to accommodate cell culture dishes increases the cost and complexity of experiments manifold times, while often suffering from restricted field of observation. Therefore, most users are forced to resort to label-dependent, end-point assays using flow cytometry.
A flow cytometry on the other hand cannot perform most of the experiments at reach with lensfree video microscopy. Obviously, measuring the orientation of cell division or monitor cell spreading is complicated if not impossible using flow cytometry. Quantifying cell adhesion or cell death also is complicated because the measurement requires the cell population to be harvested. Cell harvesting is difficult in cases where the entire population does not have same degree of attachment to the substrate, which is precisely the case during cell adhesion and cell death. Moreover, the inevitability of cell harvesting and markers perturb continuity. The only way to perform continuous measurements using flow cytometry is by conducting several measurements at different time points using cultures that are subjected to same treatment. However, the measurements cannot be called continuous since the population is not the same. For example, to the best of our knowledge, to perform the experiment shown in Fig 6c, Fig 7c,d, where the same population is monitored from cell adhesion until cell death is simply not possible with any of the flow cytometry approaches that exist today.
Overall, using our lensfree video microscopy platform, we exhaustively monitored primary human fibroblasts, hMSCs and human Osteo Sarcoma (U2OS) cells. In each case we observed thousands of cells for extended time period (20 hours to more than a week). For our experiments, we chose the cell types arbitrarily. Assessing our metrics for all cell types is beyond the scope of the manuscript. However, depending on the cell type, cell density and the application, the metrics that we report here could be updated or new metrics could be developed.
Also unlike commonly used assays that can monitor only one cell function at a given condition, our platform and associated metrics can follow all the mentioned cell functions simultaneously in the same cell culture in observation. This paves way to assess combination of cell functions hampered or enhanced by a particular scenario. In addition to cell adhesion, cell spreading, cell division and cell death, several other occurrences may also be followed in real-time. The platform is entirely non-invasive and is therefore particularly suitable for rare cell types. Also, by multiplexing several imaging sensors together, multi-well plates can be monitored39 and the throughput of the system can be dramatically increased.
Our Lensfree video microscopy prioritized simplicity and adherence to standard cell culture practices over resolution. As a result, resolution of our setup is limited. Although the setup cannot resolve fine lamellipodial and filopodial extensions (3–5 µm, Supplementary Fig. 5c), shape and the size of single cells are well reconstructed and segmented (Supplementary Fig. 5a,b). Also, we clearly demonstrated that many important measurements can be performed without requiring sub-cellular resolution or fluorescence; e.g. cell adhesion, cell division, cell motility, cell death. Our setup is able to perform measurements that are not possible with lens based microscopy or cytometer and therefore to address biological issues that were not accessible before.
Overall, with the results presented here, we demonstrate that our lensfree video microscopy and associated metrics give new perspectives to cell culture monitoring and have high potential to irreversibly change the quantification of cell behavior in cell culture conditions.
Methods
Lensfree Video Microscopy
Our lensfree video microscope consists of a 12-bit APTINA MT9P031 CMOS RGB imaging sensor with a pixel pitch of 2.2 μm, measuring 5.7 × 4.3 mm and light-emitting diode (LED) (dominating wavelength 525 nm) with a pinhole of 150 μm (Fig. 1). In a typical experiment, the lensfree video-microscope is placed inside the incubator and the culture dish (35 mm) containing the cells is placed on lensfree video-microscope (Fig. 2). Illumination is provided by the LED along with the pinhole from a distance of ~5 cm. The light scattered by the sample and the light passing directly from the source to the imaging sensor interfere to form a holographic pattern, which is recorded by the sensor and transmitted to a laptop via USB cable.
Cell Culture
Healthy donors' bone marrow (BM) samples were obtained from filters used during BM processing for allogeneic transplantation. Bone marrow samples were harvested from washed filters used during BM graft processing for allogeneic transplantation after healthy donor informed consent according to approved institutional guidelines (Assistance Publique – Hôpitaux de Paris, Paris, France). hMSCs were isolated and expanded as previously described40. Briefly, the hMSCs were cultured at the initial density of 5 × 104 cells/cm2 in Minimum Essential Medium-α (Invitrogen) supplemented with 10% Fetal Calf Serum (PAA), Glutamax-I (2 mM; Invitrogen), basic fibroblast growth factor (1 ng/mL; R&D Systems) antibiotic/antimycotic (1%, Invitrogen). After 24–48 hours, non-adherent cells were removed and medium was changed. Adherent cells were then trypsinized, harvested and cultured by seeding 5 × 103 cells/cm2. Cultures were fed every 2 to 3 days and trypsinized every 5 days. In all experiments, hMSCs were used at passage 3 to 7.
Primary cultures of human fibroblasts were prepared from skin biopsies performed on healthy donors. The cells were established in DME-Glutamax (Invitrogen) supplemented with 20% Fetal Calf Serum and used at passage 2 using standard operating procedures.
Human osteosarcoma U2OS cells were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM; Life Technologies™), supplemented with pyruvate and 10% fetal bovine serum.
To monitor cell adhesion, ~20000 cells were added to a standard 35 mm culture dishes and the dishes were immediately positioned on lens free video microscopes inside a standard incubator.
For cell death experiments involving U2OS and hMSCs, cell death was induced by forward transfection using the positive cell death phenotype control siRNA (siCellDeath = AllStars Hs Cell Death siRNA, Ref SI04381048; Qiagen) and the LipofectamineRNAiMAX transfection reagent (Life Technologies™). For transfection, LipofectamineRNAiMAX (3 µL) and siCellDeath (20 nM final concentration) were separately diluted in Opti-MEM media, combined together then added to culture dishes after 10 minutes of incubation.
Computational methods
Lensfree holographic reconstruction employed here is based on the methods described in41,42,27
Automated detection of floating and adherent cells
Automated detection of floating and adherent cells is performed on the raw image (Supplementary Video 1).
All floating cells exhibit a similar pattern corresponding to the one observed in Fig 3a at t = t0. By recognizing these patterns using normalized cross correlation function available in Matlab, floating cells are detected.
As mentioned earlier, adherent cells exhibit larger zero order gray value. Hence by applying a constant threshold based on gray value and area, adherent cells are identified.
We calculated the F1 measure (harmonic mean of precision and recall with equal weightage) in order to validate the automated detection of both floating and adherent cells. Precision or positive predictive value is defined as TP / (TP + FP), while recall or sensitivity is defined as TP / (TP + FN), where TP, FP and FN are True Positives, False Positives and False Negatives respectively. The F1 measure obtained is close to 0.95 based on 1352 cells from 10 random images from different experiments and cell types. The positive predictive value is more than 0.95 indicating that more than 95% of the detected objects correspond to cells. F1 measure was particularly used since the true negatives cannot be determined in this case.
Measurement of kurtosis and gray value
Once the cells are detected, Kurtosis is measured (using inbuilt Matlab function ‘kurtosis’) on the region of interest of 165 µm * 165 µm surrounding the detected cells. Mean zero order gray value is obtained from a smaller circular region of interest of 16 µm diameter surrounding the detected cells. Both these measurements are performed on the raw image.
Measurement of aspect-ratio
Adherent cells are segmented from the reconstructed image by applying appropriate threshold on the higher gray levels in accordance with their characteristic patterns. Then artifacts and debris, e.g. too small or too big objects, are removed from the obtained binary image by means of morphomath operators. Aspect-ratio, which is calculated on the segmented cells, is the ratio of the minor axis length to the major axis length of the ellipse which best fits the shape of the segmented cells.
Determination of longest-axis and axis of cell division
Once a dividing cell is detected based on pattern recognition10, the position of the cell in the image is used as a reference to find the same cell in previous and subsequent image frames, in order to determine the longest axis and the axis of cell division respectively. Both longest axis and cell division axis is calculated using the ‘regionprops’ function available in Matlab which calculates the orientation of the ellipse that best fits the object. In case of inability to find the longest axis or the axis of cell division, the cell is not considered for measurement.
Cell tracking
Segmented cells were tracked in subsequent temporal images using nearest neighbor linking approach with a threshold displacement (in between subsequent image frames) of 25 µm.
References
Arias, L. R., Perry, C. a. & Yang, L. Real-time electrical impedance detection of cellular activities of oral cancer cells. Biosens. Bioelectron. 25, 2225–31 (2010).
Hondroulis, E., Liu, C. & Li, C.-Z. Whole cell based electrical impedance sensing approach for a rapid nanotoxicity assay. Nanotechnology 21, 315103 (2010).
Hong, J., Kandasamy, K., Marimuthu, M., Choi, C. S. & Kim, S. Electrical cell-substrate impedance sensing as a non-invasive tool for cancer cell study. Analyst 136, 237–45 (2011).
Michaelis, S., Wegener, J. & Robelek, R. Label-free monitoring of cell-based assays: Combining impedance analysis with SPR for multiparametric cell profiling. Biosens. Bioelectron. 49C, 63–70 (2013).
Mölder, a. et al. Non-invasive, label-free cell counting and quantitative analysis of adherent cells using digital holography. J. Microsc. 232, 240–7 (2008).
Xu, W., Jericho, M. H., Meinertzhagen, I. a. & Kreuzer, H. J. Digital in-line holography of microspheres. Appl. Opt. 41, 5367–75 (2002).
Marx, V. Is super-resolution microscopy right for you? Nat. Methods 10, 1157–63 (2013).
Zheng, G., Lee, S. A., Antebi, Y., Elowitz, M. B. & Yang, C. The ePetri dish, an on-chip cell imaging platform based on subpixel perspective sweeping microscopy (SPSM). Proc. Natl. Acad. Sci. U. S. A. 108, 1–6 (2011).
Jin, G. et al. Lens-free shadow image based high-throughput continuous cell monitoring technique. Biosens. Bioelectron. 38, 126–31 (2012).
Vinjimore Kesavan, S. et al. Real-time label-free detection of dividing cells by means of lensfree video microscopy. J. Biomed. Opt. 19, 036004–1–10 (2014).
Pushkarsky, I. et al. Automated single-cell motility analysis on a chip using lensfree microscopy. Sci. Rep. 4, 4717 (2014).
Isikman, S. O., Bishara, W., Zhu, H. & Ozcan, A. Optofluidic Tomography on a Chip. Appl. Phys. Lett. 98, 161109 (2011).
Bishara, W., Zhu, H. & Ozcan, A. Holographic opto-fluidic microscopy. Opt. Express 18, 27499–510 (2010).
Khademhosseinieh, B. et al. Lensfree sensing on a microfluidic chip using plasmonic nanoapertures. Appl. Phys. Lett. 97, 221107 (2010).
Coskun, A. F. et al. A personalized food allergen testing platform on a cellphone. Lab Chip 13, 636–40 (2013).
Mudanyali, O., Oztoprak, C., Tseng, D., Erlinger, A. & Ozcan, A. Detection of waterborne parasites using field-portable and cost-effective lensfree microscopy. Lab Chip 10, 2419–23 (2010).
Coskun, A. F., Sencan, I., Su, T.-W. & Ozcan, A. Wide-field lensless fluorescent microscopy using a tapered fiber-optic faceplate on a chip. Analyst 136, 3512–8 (2011).
Sencan, I., Coskun, A. F., Sikora, U. & Ozcan, A. Spectral demultiplexing in holographic and fluorescent on-chip microscopy. Sci. Rep. 4, 3760 (2014).
Coskun, A. F., Sencan, I., Su, T.-W. & Ozcan, A. Lensfree fluorescent on-chip imaging of transgenic Caenorhabditis elegans over an ultra-wide field-of-view. PLoS One 6, e15955 (2011).
Isikman, S. O. G. A. M. B. W. O. S. T.-W. O. A. Lensfree computational microscopy tools for cell and tissue imaging at the point-of-care and in low-resource settings. Anal. Cell Pathol.(Amst). 35, 229–247 (2012).
Su, T.-W., Seo, S., Erlinger, A. & Ozcan, A. High-throughput lensfree imaging and characterization of a heterogeneous cell solution on a chip. Biotechnol. Bioeng. 102, 856–68 (2009).
Weidling, J., Isikman, S. O., Greenbaum, A., Ozcan, A. & Botvinick, E. Lens-free computational imaging of capillary morphogenesis within three-dimensional substrates. J. Biomed. Opt. 17, 126018 (2012).
Su, T.-W., Erlinger, A., Tseng, D. & Ozcan, A. Compact and light-weight automated semen analysis platform using lensfree on-chip microscopy. Analytical chemistry. 82, 8307–12 (2010).
Seo, S. et al. High-Throughput Lens-Free Blood Analysis on a Chip. Analytical chemistry. 82, 4621–4627 (2010).
Ozcan, A. & Demirci, U. Ultra wide-field lens-free monitoring of cells on-chip. Lab Chip 8, 98–106 (2008).
Navruz, I. et al. Smart-phone based computational microscopy using multi-frame contact imaging on a fiber-optic array. Lab Chip 13, 4015–23 (2013).
Kim, S. B. et al. A cell-based biosensor for real-time detection of cardiotoxicity using lensfree imaging. Lab Chip 11, 1801–7 (2011).
Cuvelier, D. et al. The universal dynamics of cell spreading. Curr. Biol. 17, 694–9 (2007).
McGrath, J. L. Cell spreading: the power to simplify. Curr. Biol. 17, R357–8 (2007).
Burmeister, J. S., Olivier, L. a., Reichert, W. M. & Truskey, G. a. Application of total internal reflection fluorescence microscopy to study cell adhesion to biomaterials. Biomaterials 19, 307–25 (1998).
Hug, T. S., Prenosil, J. E. & Morbidelli, M. Optical waveguide lightmode spectroscopy as a new method to study adhesion of anchorage-dependent cells as an indicator of metabolic state. Biosens. Bioelectron. 16, 865–74 (2001).
Ramsden, J. J., Li, S. Y., Heinzle, E. & Prenosil, J. E. Optical method for measurement of number and shape of attached cells in real time. Cytometry 19, 97–102 (1995).
Sund, S. E. & Axelrod, D. Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleaching. Biophys. J. 79, 1655–69 (2000).
Gillies, T. E. & Cabernard, C. Cell division orientation in animals. Curr. Biol. 21, R599–609 (2011).
Minc, N., Burgess, D. & Chang, F. Influence of cell geometry on division-plane positioning. Cell 144, 414–26 (2011).
Théry, M. & Bornens, M. Cell shape and cell division. Curr. Opin. Cell Biol. 18, 648–57 (2006).
Théry, M. et al. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol. 7, 947–53 (2005).
Kepp, O., Galluzzi, L., Lipinski, M., Yuan, J. & Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 10, 221–37 (2011).
Haguet, V., Obeïd, P., Griffin, R., Freida, D. & Gidrol, X. Parallelized lensfree time-lapse microscopy. Conf. NanoBioEurope, p. 30, Toulouse, Fr. June 10–12 (2013).
Arnulf, B. et al. Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia 21, 158–63 (2007).
Mudanyali, O. et al. Compact, light-weight and cost-effective microscope based on lensless incoherent holography for telemedicine applications. Lab Chip 10, 1417–1428 (2010).
Allen, L. J. & Oxley, M. P. Phase retrieval from series of images obtained by defocus variation. Opt. Commun. 199, 65–75 (2001).
Acknowledgements
Authors wish to thank T. Bordy, JG. Coutard, S. Morel for their help with the lensfree video microscopy platform. We thank L. Herve for his help with Matlab programming. We wish to thank R. Sauze for the image acquisition software and H. Grateau for the fabrication of the lensfree video microscopy setup.
Author information
Authors and Affiliations
Contributions
S.V.K., C.A., B.D.W., N.D., S.L.S., E.S., D.F., X.G. designed and performed the experiments. C.A., F.M., O.C. performed holographic reconstruction and segmentation. S.V.K., C.A., F.M., performed image analysis. S.V.K., C.A., F.M. wrote the manuscript. C.A., B.C., X.G., J.M.D. supervised the projects. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Video 1
Supplementary Information
Supplementary Video 2
Supplementary Information
Supplementary Video 3
Supplementary Information
Supplementary Video 4
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Kesavan, S., Momey, F., Cioni, O. et al. High-throughput monitoring of major cell functions by means of lensfree video microscopy. Sci Rep 4, 5942 (2014). https://doi.org/10.1038/srep05942
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05942
This article is cited by
-
A novel feature for monitoring the enzymatic harvesting process of adherent cell cultures based on lens-free imaging
Scientific Reports (2022)
-
Contact guidance as a consequence of coupled morphological evolution and motility of adherent cells
Biomechanics and Modeling in Mechanobiology (2022)
-
Nonmechanical parfocal and autofocus features based on wave propagation distribution in lensfree holographic microscopy
Scientific Reports (2021)
-
Low-cost calcium fluorometry for long-term nanoparticle studies in living cells
Scientific Reports (2020)
-
The physics of cell-size regulation across timescales
Nature Physics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
