
Abstract
The abnormal growth of vascular smooth muscle cells (VSMCs) is considered a critical pathogenic process in inflammatory vascular diseases. We have previously demonstrated that protein phosphatase 2 A (PP2A)-mediated NF-κB dephosphorylation contributes to the anti-inflammatory properties of andrographolide, a novel NF-κB inhibitor. In this study, we investigated whether andrographolide causes apoptosis and characterized its apoptotic mechanisms in rat VSMCs. Andrographolide activated the p38 mitogen-activated protein kinase (p38MAPK), leading to p53 phosphorylation. Phosphorylated p53 subsequently transactivated the expression of Bax, a pro-apoptotic protein. Transfection with pp2a small interfering RNA (siRNA) suppressed andrographolide-induced p38MAPK activation, p53 phosphorylation and caspase 3 activation. Andrographolide also activated the Src homology 1 domain-containing protein tyrosine phosphatase (SHP-1) and induced PP2A dephosphorylation, both of which were inhibited by the SHP-1 inhibitor sodium stibogluconate (SSG) or shp-1 siRNA. SSG or shp-1 siRNA prevented andrographolide-induced apoptosis. These results suggest that andrographolide activates the PP2A-p38MAPK-p53-Bax cascade, causing mitochondrial dysfunction and VSMC death through an SHP-1-dependent mechanism.
Similar content being viewed by others


Introduction
Vascular restenosis in intimal hyperplasia remains a major problem for long-term patency of angioplasty and stent placement in the treatment of obstructive arterial diseases. Therefore, the development of novel therapeutic agents for patients with vascular restenosis remains a major research priority1. The activation of monocytes, macrophages and inflammation and the proliferation of vascular smooth muscle cells (VSMCs) are thought to be crucial in the development of intimal hyperplasia2,3,4. Pharmacological approaches to alleviate inflammation or to elicit VSMC death may provide new strategies for managing vascular restenosis and obstructive arterial diseases.
Preparations containing components of Andrographis paniculata have been widely used in traditional Asian medicine to treat upper respiratory tract infections, fever, diarrhea, rheumatoid arthritis and laryngitis5,6. A recent clinical trial showed that an A. paniculata-based treatment relieved the symptoms of rheumatoid arthritis7. Andrographolide is the most active constituent in extracts of the leaves of A. paniculata and may thus have immunomodulatory and anti-inflammatory activities. In addition, andrographolide has exhibited anti-cancer properties8,9. Andrographolide was shown to inhibit tumor growth by inducing cell cycle arrest10,11 or apoptosis12,13 in various types of cancer cells. Andrographolide also suppresses v-Src transformation and sensitizes cancer cells to apoptosis mediated by the tumor necrosis factor-related apoptosis-inducing ligand14. The underlying mechanisms by which andrographolide exhibits these various activities remains unclear.
Studies have demonstrated that the anti-inflammatory properties of andrographolide can be attributed to the inhibition of the nuclear factor (NF)-κB pathway8,9,15. We have previously demonstrated that andrographolide enhances the dephosphorylation of NF-κB subunit p65 Ser-536 through the activation of protein phosphatase 2 A (PP2A) in rat VSMCs exposed to inflammatory stimuli16. However, although andrographolide has demonstrated anti-proliferative and apoptotic effects on various types of cancer cells, whether it induces apoptosis in VSMCs is not known. Furthermore, although PP2A appears to mediate the anti-inflammatory activity of andrographolide, the role of PP2A in andrographolide-induced apoptosis remains unclear.
Reversible protein phosphorylation catalyzed by protein kinases and protein phosphatases regulates various cellular processes, including inflammation and apoptosis17,18. Recent studies have highlighted a major role of serine/threonine protein phosphatases, including that of PP2A in apoptosis19,20. The PP2A is a member of the ceramide-activated protein phosphatases (CAPPs) that is composed of regulatory subunits A and B and a catalytic subunit C21. The activity of PP2A is regulated by interactions of regulatory subunits22 or by post-translational modifications, including methylation23 and phosphorylation21,24. The methylation of PP2A at Leu-309 activates PP2A23 and phosphorylation at Tyr-307 suppresses PP2A catalytic activity21.
The role of PP2A in cell apoptosis has been shown to involve the upregulation of Bim20 or Bax19, which are both pro-apoptotic members of the Bcl-2 family. The Bcl-2 family includes proteins with anti-apoptotic or pro-apoptotic properties that regulate the intrinsic (mitochondrial) apoptotic pathway25. Both Bim and Bax independently interfere with the anti-apoptotic functions of other Bcl-2 family members26. The tumor suppressor protein p53 is a transcription factor that has been shown to induce apoptosis by causing mitochondrial dysfunction through the transactivation of Bax expression19,27. The p53 protein is activated primarily by phosphorylation of Ser-1528.
We have previously shown that the p38 mitogen-activated protein kinase (MAPK) mediates the phosphorylation of Ser-15 in p53 and the subsequent expression of Bax during apoptosis in cerebral endothelial cells21,29. Because pro-apoptotic Bcl-2 family members, such as Bax, are believed to contribute to the apoptotic actions of andrographolide12,13, we investigated whether p53 contributes to andrographolide-induced apoptosis in VSMCs. Our findings support the contention that the activation of a PP2A-p38MAPK-p53-Bax pathway mediated by the Src homology 1 domain-containing protein tyrosine phosphatase (SHP-1) contributes to andrographolide-induced apoptosis in VSMCs.
Results
Andrographolide induced cell apoptosis in rat VSMCs
An MTT assay was used to examine the effects of andrographolide on cell viability in rat VSMCs. As shown in Figure 1a, treatment with 50 μM andrographolide significantly decreased cell viability by 20.8% ± 4.3% and 75.8% ± 5.2% after 24 h and 48 h, respectively (n = 4). Treatment with 20 or 50 μM andrographolide induced changes in cellular morphology, with cells becoming round and shrunken (Figure 1b). We also determined whether andrographolide induced VSMC death by apoptosis. In healthy cells, the phosphatidylserine (PS) groups in the plasma membrane are directed toward the inside of the cell. However, the PS groups are exposed to the environment upon apoptosis. We used supravital exposure to propidium iodide and annexin V double labeling for the detection of apoptosis. Compared with the vehicle-treated cells (control group, Figure 1c, panel i), the cells treated with 20 μM (Figure 1c, panel ii) or 50 μM (Figure 1c, panel iii) andrographolide showed a higher proportion of annexin V labeling, compared with control cells and displayed 2 levels of labeling. The annexin V+PI− cells (lower right quadrant) represented early apoptotic cells and the annexin V+PI+ cells (upper right quadrant) represented advanced apoptotic cells and/or necrotic cells. These results suggest that andrographolide induced apoptosis in the rat aortic VSMCs.

Effects of andrographolide on cell apoptosis in rat VSMCs.
(a) Cells were treated with andrographolide at indicated concentrations for 24 h or 48 h and cell viability was then determined by MTT assay. Each column represents the mean ± S.E.M. of at least four independent experiments. (**, P < 0.01 and ***, P < 0.001, compared with the vehicle-treated control group). (b) Photomicrographs showing the effect of treatment with vehicle (i) or 20 μM (ii) or 50 μM (iii) andrographolide for 48 h. (magnification × 100). (c) Cells were treated with andrographolide at indicated concentrations for 48 h. Cells were then stained with annexin V and propidium iodide for 15 min. The percentage of apoptotic cells was then analyzed by flow-cytometric analysis. Results shown are representative of four independent experiments.
Andrographolide activated mitochondrial apoptotic pathway in rat VSMCs
Mitochondria are thought to be the major pathway for apoptosis30,31. The pro-apoptotic Bcl-2 family member Bax plays a crucial role in the mitochondrial control of apoptosis32. To determine whether the mitochondrial apoptotic signaling pathway was involved in the andrographolide-induced apoptosis of rat aortic VSMCs, the expression profile of Bax was examined. As shown in Figure 2a, treatment with 20 or 50 μM andrographolide for 48 h significantly induced Bax expression by 1.6 ± 0.1 fold and 2.4 ± 0.1 fold, respectively (n = 3).

Effects of andrographolide on the intrinsic mitochondrial pathway in rat VSMCs.
(a) Cells were treated with andrographolide at indicated concentrations for 48 h. The extent of Bax level was then determined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of three independent experiments. The full-length blot is presented in Supplementary Figure 1a. (b) Cells were treated with andrographolide at indicated concentrations for 48 h. The change of mitochondria membrane potential in JC-1-stained cells was then determined by flowcytometry. Cells were treated with andrographolide at indicated concentrations for 48 h. Results shown are representative of six independent experiments. The extent of cleavage caspase 9 (c) and cleavage caspase 3 (d) were then determined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of four independent experiments. The full-length blots are presented in Supplementary Figure 1b and 1c. (*, P < 0.05, **, P < 0.01 and ***, P < 0.001, compared with the control group).
The change of mitochondria membrane potential (Δψm) is an important factor in inducing apoptosis32. We used the selective mitochondrial uptake of the JC-1 dye to examine the Δψm following treatment with andrographolide. When the mitochondrial membrane potential decreases, the JC-1 dye selectively enters the mitochondria matrix and the color of JC-1 fluorescence changes from red to green. The healthy living cells maintain a high mitochondrial membrane potential and JC-1 spontaneously forms the red J-aggregates, whereas apoptotic cells have a low potential, resulting in the formation of the green JC-1 monomers. As shown in Figure 2b, treatment with 20 or 50 μM andrographolide induced a negative Δψm in rat VSMCs, compared with the vehicle-treated control group.
Because an increase in the permeability of the outer mitochondria membrane leads to the release of several apoptotic factors, such as cytochrome c, that activate caspases which mediate apoptosis, we examined the activation of caspase-9 and caspase-3 in andrographolide-treated VSMCs. As shown in Figure 2c, the level of the cleaved, activated form of caspase-9 increased following treatment with andrographolide for 48 h (n = 4). The andrographolide treatment also caused an increase in the level of the cleaved, activated form of caspase-3 (n = 4) (Figure 2d) These observations indicate that andrographolide induces mitochondrial apoptosis in rat VSMCs.
Andrographolide-induced p53 phosphorylation and activation in rat VSMCs is mediated by p38MAPK
The transcription factor p53 has been shown to induce apoptosis by causing mitochondrial dysfunction through the transactivation of Bax33,34. The phosphorylation of p53 at Ser-15 has been shown to contribute to its activation28. To explore the role of p53 in andrographolide-induced VSMC apoptosis, we examined the effect of andrographolide on the phosphorylation of Ser-15 in p53. As shown in Figure 3a, treatment with andrographolide increased p53 Ser-15 phosphorylation. Following andrographolide treatment, detectable phosphorylation began at 10 min, peaked at 20 min and declined to basal levels after 30 min (n = 4).

Involvement of p38MAPK in andrographolide-induced p53 phosphorylation in rat VSMCs.
Cells were treated with 50 μM andrographolide for the indicated periods. The extent of p53 phosphorylation (a) and p38 phosphorylation (b) were then determined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of at least three independent experiments. The full-length blots are presented in Supplementary Figure 2a and 2b (c) Cells were pretreated with vehicle, 10 μM SB203580, or 1 μM p38MAPK inhibitor III for 30 min followed by the treatment with 50 μM andrographolide for another 48 h. Cell viability was then determined by MTT assay. Each column represents the mean ± S.E.M. of four independent experiments. (d) Cells were transiently transfected with PG-13-luc and Renilla-luc for 48 h. After transfection, cells were pretreated with vehicle or SB203580 (10 μM) for 30 min followed by the treatment with andrographolide (50 μM) for another 24 h. The PG13-luciferase activity was then determined. Each column represents the mean ± S.E.M. of four independent experiments. (e) Cells were pretreated with vehicle or 10 μM SB203580 for 30 min followed by the treatment with 50 μM andrographolide for another 48 h. The extent of Bax level was then determined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of three independent experiments. The full-length blot is presented in Supplementary Figure 2c. (*, P < 0.05, **, P < 0.01 and ***, P < 0.001, compared with the control group; and #, P < 0.05 and ##, P < 0.01, compared with the group treated with andrographolide alone).
We have previously demonstrated that p38MAPK signaling is responsible for p53 Ser-15 phosphorylation in cerebral endothelial cells19 and colon cancer cells35. Thus, we examined whether p38MAPK phosphorylation is altered in rat VSMCs treated with andrographolide. As shown in Figure 3b, treatment with andrographolide induced p38MAPK phosphorylation (n = 3). We also examined whether p38MAPK contributes to andrographolide-induced VSMC death using the p38MAPK signaling inhibitors, SB203580 and p38MAPK inhibitor III. Pretreatment with SB203580 (10 μM) or p38MAPK inhibitor III (1 μM) significantly restored andrographolide-decreased cell viability (n = 4) (Figure 3c).
We also used a PGl3-Luc reporter construct that contained a p53 DNA-binding site linked to a basal promoter that controls the expression of a luciferase reporter gene36 to examine whether p53 transactivation increases in cells exposed to andrographolide. As shown in Figure 3d, cells treated with 50 μM andrographolide for 24 h exhibited an increase in PG13-luciferase activity of 3.5 ± 0.5 fold (n = 5). The pretreatment of cells with SB203580 inhibited the andrographolide-induced increase in PG13-luciferase activity (Figure 3d) and suppressed Bax expression (Figure 3e) in rat VSMCs exposed to andrographolide. These results suggest that p38MAPK may mediate p53 activation, Bax expression and apoptosis in rat VSMCs treated with andrographolide.
PP2A mediates andrographolide-activated p38MAPK-p53 cascade in rat VSMCs
We investigated the signaling molecules that may contribute to the andrographolide-induced activation of the p38MAPK-p53-Bax cascade in rat VSMCs. We recently demonstrated that andrographolide increased the protein phosphatase activity of PP2A in rat VSMCs16. In addition, PP2A activates p5319 and PP2A has been shown to contribute to various cellular responses through the regulation of ASK1, p38MAPK and JNK18,19. We examined the effects of pp2a siRNA in rat VSMCs exposed to andrographolide to determine the relationship between PP2A activation and the p38MAPK-p53 cascade. Transfection of rat VSMCs with pp2a siRNA significantly inhibited andrographolide-increased p38 MAPK (Figure 4a) and p53 (Figure 4b) phosphorylation. The pp2a siRNA also reduced the andrographolide-induced activation of caspase-3 (Figure 4c). Furthermore, pp2a siRNA significantly restored andrographolide-decreased cell viability in rat VSMCs (Figure 4d) (n = 4). siRNA experiments also revealed that pp2a siRNA suppressed the basal level of PP2A-C (Figure 4e). These results suggest that PP2A may be responsible for the andrographolide-induced activation of the p38MAPK-p53 cascade in rat VSMCs.

PP2A is involved in andrographolide-induced p38MAPK, p53 and caspase-3 activation in rat VSMCs.
Cells were transiently transfected with pp2a siRNA for 48 h followed by the treatment with andrographolide for another 10 min (a and b) or 48 h (c). The extent of p38 phosphorylation (a), p53 phosphorylation (b) and cleavaged caspase-3 (c) were then examined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of at least three independent experiments. The full-length blots are presented in Supplementary Figure 3a, 3b and 3c. (d) Cells were transiently transfected with pp2a siRNA for 48 h followed by the treatment with andrographolide for another 48 h. Cell viability was then determined by MTT assay. Each column represents the mean ± S.E.M. of four independent experiments. (e) Cells were transiently transfected with pp2a siRNA for 48 h followed by the treatment with andrographolide for another 10 min. The extent of PP2A catalytic subunit (PP2A-C) was then examined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of three independent experiments. The full-length blots are presented in Supplementary Figure 3d. (*, P < 0.05 and ***, P < 0.001, compared with the control group; and #, P < 0.05, ##, P < 0.01 and ###, P < 0.001, compared with the group treated with andrographolide alone). The full-length blots are presented in Supplementary Figure 3d.
Andrographolide activated SHP-1 in rat VSMCs
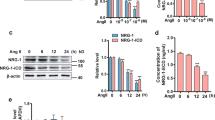
The precise mechanism involved in the andrographolide-induced activation of PP2A in rat VSMCs remains unclear. We previously demonstrated that PP2A is activated by the neutrosphingomyelinase-ceramide cascade in rat VSMCs exposed to andrographolide16. However, PP2A activity may also be regulated by methylation or phosphorylation21,23,24. Thus, we examined whether the methylation or phosphorylation of PP2A-C is altered by andrographolide. As shown in Figure 5a, treatment with andrographolide had no effect on the level of demethylated PP2A-C (DM-PP2A-C, inactive form). However, treatment with andrographolide for 10 to 30 min caused a significant decrease in the phosphorylation of Tyr-307 in PP2A (Figure 5a), whereas the total level of PP2A-C was not changed. These results indicate that andrographolide may activate PP2A by regulating PP2A-C Tyr-307 phosphorylation.

SHP-1 is involved in andrographolide-induced PP2A dephosphorylation in rat VSMCs.
(a)Cells were treated with 50 μM andrographolide for the indicated time periods. Cells were then harvested and PP2A-C demethylation and Tyr-307 phosphorylation were examined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of four independent experiments. The full-length blots are presented in Supplementary Figure 4a. Cells were pretreated with vehicle or 10 μM SSG for 30 min followed by the treatment with 50 μM andrographolide for another 30 min (b) or 48 h (c). The extent of PP2A-C Tyr-307 phosphorylation was them examined by immunoblotting (b). Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of three independent experiments. The full-length blot is presented in Supplementary Figure 4b. Cell viability was determined by MTT assay (c). Each column represents the mean ± S.E.M. of four independent experiments. (d) Cells were transiently transfected with shp-1 siRNA for 48 h followed by the treatment with andrographolide for another 30 min. The extent of PP2A-C Tyr-307 phosphorylation was them examined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of four independent experiments. The full-length blot is presented in Supplementary Figure 4c. (e) Cells were transiently transfected with shp-1 siRNA for 48 h followed by the treatment with andrographolide for another 48h. Cell viability was then determined by MTT assay. Each column represents the mean ± S.E.M. of four independent experiments. (f) Cells were treated as described in (d). The extent of SHP-1 was then examined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of four independent experiments. The full-length blot is presented in Supplementary Figure 4d. (*, P < 0.05, **, P < 0.01 and ***, P < 0.001, compared with the control group; and #, P < 0.05 and ###, P < 0.001, compared with the group treated with andrographolide alone).
We investigated the underlying mechanism by which andrographolide induces PP2A Tyr-307 dephosphorylation. Andrographolide may activate a protein tyrosine phosphatase that dephosphorylates PP2A-C Tyr-307, leading to PP2A activation. Activation of SHP-1 was recently shown to play an important role in the vascular protective effects of estrogen in rat VSMCs37. We, therefore, explored whether SHP-1 is involved in PP2A-C dephosphorylation. Pretreatment with the selective SHP-1 inhibitor SSG38 reduced PP2A-C Tyr-307 dephosphorylation in rat VSMCs treated with andrographolide (Figure 5b). Pretreatment with SSG also inhibited the andrographolide-induced reduction in cell viability in rat VSMCs (Figure 5c). To further confirm more specifically that andrographolide-induced PP2A-C Tyr-307 dephosphorylation was mediated by SHP-1, shp-1 siRNA oligonucleotide was used. As shown in Figure 5d, transfection of rat VSMCs with shp-1 siRNA significantly reduced andrographolide-induced PP2A-C Tyr-307 dephosphorylation. shp-1 siRNA also restored andrographolide-decreased cell viability in rat VSMCs (Figure 5e). Furthermore, siRNA experiments revealed that shp-1 siRNA suppressed the basal level of SHP-1 (Figure 5f).
We also examined whether andrographolide activates SHP-1 in rat VSMCs. A member of a family of non-receptor protein tyrosine phosphatases (PTPs), SHP-1 contains 2 SH2 domains and is activated by the phosphorylation of Tyr-5644. As shown in Figure 6a, andrographolide increased the phosphorylation of Tyr-564 in SHP-1. Andrographolide also significantly increased the phosphatase activity of SHP-1 (Figure 6b). Consistent with the inhibitory effects of SSG, a reduction in SHP-1 enzyme activity was observed in rat VSMCs exposed to andrographolide in the presence of SSG (Figure 6b). Moreover, the andrographolide-induced PP2A activation was significantly suppressed by SSG (Figure 6c). These results suggest that SHP-1 may mediate andrographolide-induced dephosphorylation of PP2A-C Tyr-307 in rat VSMCs, which leads to the activation of the p38MAPK-p53-Bax cascade and subsequent apoptosis.

Effects of andrographolide on SHP-1 and PP2A activity in rat VSMCs.
(a) Cells were treated with 50 μM andrographolide for the indicated time periods. The extent of SHP-1 phosphorylation was then determined by immunoblotting. Compiled results are shown at the bottom of the chart. Each column represents the mean ± S.E.M. of four independent experiments. Cells were pretreated with vehicle or 10 μM SSG for 30 min followed by the treatment with 50 μM andrographolide for another 10 min. The enzyme activity of SHP-1 (b) and PP2A (c) were then determined. Compiled results are shown at the bottom of each chart. Each column represents the mean ± S.E.M. of at least four independent experiments. The full-length blot is presented in Supplementary Figure 5. (*, P < 0.05 and **, P < 0.01, compared with the control group; and #, P < 0.05, compared with the group treated with andrographolide alone). IP: immunoprecipitation. IB: immunoblotting.
Discussion
Andrographolide, the principal bioactive compound in extracts of the leaves of A. paniculata, is a novel NF-κB inhibitor8,9,15. Our previously study showed that the PP2A-mediated dephosphorylation of NF-κB contributes to the protective anti-inflammatory properties of andrographolide in rat VSMCs. We also showed that andrographolide inhibited the formation of the neointima in a rat carotid balloon angioplasty model16. Our current findings demonstrate that andrographolide activates the SHP-1-PP2A-p38MAPK-p53 cascade, which induces Bax expression, mitochondria dysfunction, casepase-3 activation and subsequent apoptosis in VSMCs.
We established previously that ceramide activation of the CAPP family member PP2A may be involved in andrographolide-induced p65 dephosphorylation in VSMCs16. In addition, PP2A activity is also regulated by phosphorylation21,24. Phosphorylation of Tyr-307 in PP2A-C by Src family tyrosine kinases is required to maintain the inactivation of PP2A24. The signaling pathways that are required for andrographolide-induced activation of PP2A in VSMCs are not known. In our present study, we demonstrated that SSG, a specific inhibitor of SHP-1, inhibited both the andrographolide-induced dephosphorylation of Tyr-307 in PP2A-C and the activation of PP2A. Andrographolide also decreased cell viability in VSMCs through a SHP-1-dependent mechanism. These findings suggest that SHP-1 may play a pivotal role in the dephosphorylation of PP2A-C, the activation of PP2A and subsequent signaling events in VSMCs exposed to andrographolide.
Although closely related in structure, the roles of SHP-1 and SHP-2 in cells are distinct. The SHP-1 protein regulates signaling in a negative manner39 by suppressing the phosphorylation of proteins, whereas SHP-2 leads to cellular activation40. Despite their structural similarity, differences in their structures are likely related to their effects on phosphatase activity. However, the underlying mechanisms through which SHP-1 and SHP-2 regulate phosphatases remains unclear. Since SHP-1 is reported to be associated with the downregulation of Src41,42, future studies of its role in the dephosphorylation of PP2A-C through Src inactivation are warranted.
The activation of PP2A has been shown to induce apoptosis by stimulating downstream signaling events, including the activation of p38MAPK19. Whether the p38MAPK signaling pathway contributes to andrographolide-induced apoptosis in VSMCs has not been demonstrated previously. Our current results show that p38MAPK was activated by andrographolide and that its activation was causally related to apoptosis in VSMCs treated with andrographolide. In addition, we found that the andrographolide-induced activation of p38MAPK and subsequent apoptosis in VSMCs were mediated by PP2A. These findings are consistent with the observation that SB203580 and pp2a siRNA inhibit both the activation of p38 MAPK and the induction of apoptosis in VSMCs treated with andrographolide. In addition to p38MAPK signaling, we recently demonstrated that ceramide-p47phox-ROS signaling cascade contributes to andrographolide-induced VSMC apoptosis43. Bao et al.44 also indicated that andrographolide causes apoptosis via inactivation Akt signaling. The link between these apoptotic pathways in VSMCs exposed to andrographolide remains to be established. These observations explain, at least in part, why blocking the p38MAPK signaling could not completely abolish andrographolide-induced VSMC apoptosis.
Increased oxidative stress may also contribute to andrographolide-induced apoptosis in VSMCs. It has been shown that mediators of oxidative stress, such as H2O2, may activate PP2A-like protein phosphatases, resulting in apoptosis45. We recently found that andrographolide increased the level of H2O2 in VSMCs43. These observations suggest that andrographolide-induced oxidative stress may be critical in activating PP2A-p38MAPK-mediated apoptotic signaling in VSMCs.
The pro-apoptotic protein Bax causes apoptosis by disrupting mitochondrial integrity46. Yang et al. demonstrated that andrographolide induces the expression of Bax, activates caspases and stimulates apoptosis in lymphoma cells47. Although the signaling events involved in andrographolide-induced Bax expression in VSMCs have not been delineated, p53 is likely to be involved. The expression of Bax is induced by p53 in response to selected stress signals48 and p53 activity is regulated by phosphorylation16. However, whether andrographolide alters the p53 phosphorylation has not been previously demonstrated.
Our current results show that treatment of VSMCs with andrographolide caused p53 Ser-15 phosphorylation. The phosphorylation of p53 by p38MAPK contributes to multiple proposed apoptosis mechanisms19,49. The expression and activation of pro-apoptotic function of Bax is also induced by PP2A26. These findings are consistent with our data which show that both the pp2a siRNA and SB203580 inhibited p53 phosphorylation, Bax expression and apoptosis in VSMCs treated with andrographolide. Thus, andrographolide may activate the SHP-1-PP2A-p38MAPK cascade in VSMCs, causing subsequent p53 phosphorylation and apoptosis.
Andrographolide has also been shown to dephosphorylate the NF-κB p65 subunit, which results in the inactivation of NF-κB signaling46. The expression of a number of anti-apoptosis genes are regulated by NF-κB50. It is therefore possible that transcription factors other than p53, including NF-κB, may also contribute to andrographolide-induced apoptosis in VSMCs. The link between these transcription factors with regard to the apoptotic properties of andrographolide remains to be established.
In conclusion, the results of our present study demonstrate for the first time that andrographolide induces apoptosis in VSMCs. The apoptotic properties of andrographolide involve the activation of the SHP-1-PP2A-p38MAPK signaling cascade, p53 activation and Bax expression.
Methods
Reagents
The Dulbecco modified Eagle medium (DMEM), optiMEM, trypsin (0.25%), L-glutamine, penicillin, streptomycin, fetal bovine serum (FBS) and the Dead Cell Apoptosis Kit with Annexin V Alexa Fluor 488 and PI were purchased from Gibco (Gaithersburg, MD, USA). The andrographolide (≥98% purity), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), SB203580, ethylenediaminetetraacetic acid (EDTA) and sodium stibogluconate (SSG) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The p38MAPK inhibitor III was purchased from Millipore (Billerica, MA, USA). The Turbofect in vitro transfection reagent was purchased from Upstate Biotechnology (Lake Placid, NY, USA). The PG13-luc plasmid (Addgene, Cambridge, MA, USA) containing the p53 binding site was kindly provided by Dr. Bert Vogelstein36. The Renilla-luc and Dual-Glo luciferase assay systems were purchased from Promega (Madison, WI, USA). The 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine chloride (JC-1) was purchased from BioVision (Mountain View, CA, USA). The Hybond-P polyvinylidene difluoride (PVDF) membrane, enhanced chemiluminescence (ECL) detection reagent were purchased from Amersham (Buckinghamshire, UK). Andrographolide was dissolved in 0.1% dimethyl sulfoxide (DMSO) and stored at 4°C.
Antibodies
The anti-caspase-9 and anti-caspase-3 monoclonal antibodies (mAbs), anti-Bax polyclonal antibody (pAb), anti-phospho-p53 pAb, anti-p53 mAb, anti-phospho-p38 MAPK mAb, anti-p38 MAPK mAb and anti-phospho-SHP-1 mAb were purchased from Cell Signaling (Beverly, MA, USA). The anti-phospho-PP2A-C mAb, anti-demethylated-PP2A-C mAb and anti-SHP-1 pAb were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The anti-PP2A-C pAb was purchased from Genetex (San Antonio, TX, USA) and the anti-α-tubulin mAb was purchased from NeoMarkers (Fremont, CA, USA). The horseradish peroxidase (HRP)-conjugated donkey anti-rabbit immunoglobulin G (IgG) and sheep anti-mouse IgG were purchased from Amersham.
Rat aortic SMC primary culture
Our animal protocols were approved by the Animal Care and Use Committee of Taipei Medical University. Male Wistar rats weighing 250 to 300 g were purchased from BioLASCO (Taipei, Taiwan) and were handled in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The VSMCs were harvested from male Wistar rats. Thoracic aortas from Wistar rats were removed and stripped of endothelium and adventitia. The VSMCs were obtained by a modification of the combined collagenase and elastase digestion method51. The harvested VSMCs were grown in DMEM supplemented with 20 mM HEPES, 10% FBS, 1% penicillin/streptomycin and 2 mM L-glutamine at 37°C in a humidified atmosphere of 5% CO2. The growth medium was changed every 2 to 3 days until cells were confluent. The growth medium was removed and the monolayer was rinsed with phosphate-buffered saline (PBS). A trypsin-EDTA solution was added and the monolayer was incubated at 37°C for 2 min. The culture dishes were observed under a phase-contrast microscope until the cells had detached. Cells were removed with 10 ml of DMEM and centrifuged at 900 × g for 7 min before being resuspended in DMEM. The cells from passages 4 to 8 were used in all of our experiments. Primary cultured rat aortic VSMCs showed the typical “hills and valleys” growth pattern and the expression of α-smooth muscle actin was confirmed (data not shown).
Cell viability assay
The VSMCs (2 × 104 cells/well) were seeded in 24-well plates and cultured in DMEM containing 10% FBS for 24 h. The VSMCs were pretreated with 20 or 50 μM andrographolide or an identical volume of solvent control (0.1% DMSO) for 24 or 48 h. Cell morphology was evaluated by phase contrast microscopy without preliminary fixation. The micrographs were recorded using a Nikon phase-contrast microscope (Tokyo, Japan). Cell viability was quantified using a colorimetric assay that measured the mitochondrial activity of the viable cells based on the reduction of MTT as previously described52. The cell number index was calculated as the ratio of the absorbance of treated cells to that of the control cells (treated/control) multiplied by 100%.
Apoptosis assays in rat VSMCs
The apoptosis assay was performed using annexin V/propidium iodide according to a method described previously53. The VSMCs (5 × 105 cells/dish) were incubated with 20 or 50 μM andrographolide or a solvent control. After 48 h, the cells were diluted to 1 × 106 cells/ml in annexin binding buffer and incubated for 15 min with the Alexa Fluor 647 annexin V conjugate. The necrotic cells were identified with propidium iodide staining and the cells were analyzed on a Coulter Epics XL flow cytometry (Beckman Coulter, Fullerton, CA, USA). The data were collected from 10000 cells per experimental group. All experiments were repeated at least 4 times to ensure reproducibility.
Mitochondrial membrane potential assay
The change in the mitochondrial membrane potential (ψm) was determined using JC-1 as described previously54. The VSMCs (5 × 105 cells/dish) were incubated with 20 or 50 μM andrographolide or a solvent control. After 24 h, the VSMCs were combined with 2 μg/ml JC-1 in DMSO for 20 min. The final volume was brought to 1 ml for immediate analysis using a Coulter Epics XL flow cytometer. The selective entry of the lipophilic cationic fluorescence dye JC-1 from the cytosol into the mitochondria shifts the fluorescence of cells from red (JC-1 aggregates, FL3) to green (JC-1 monomers, FL1) and the loss of the selective uptake of JC-1 represents the Δψm.
Immunoblotting
Immunoblotting was performed as described previously51. The VSMCs (5 × 105 cells/dish) were treated with 20 or 50 μM andrographolide or a solvent control for 20 min and the proteins were extracted with lysis buffer. The lysates were centrifuged, the supernatant was collected and 50 μg of soluble protein was analyzed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The separated proteins were electrophoretically transferred to a 0.45-μm PVDF membrane by semidry transfer (Bio-Rad, Hercules, CA, USA). Blots were blocked with 10 mM Tris-base, 100 mM NaCl and 0.01% Tween 20 (TBST) containing 5% bovine serum albumin for 1 h, before being probed with the various primary antibodies. Membranes were incubated with HRP-anti-mouse IgG or anti-rabbit IgG (diluted 1:3000 in TBST) for 1 h. The immunoreactive bands were detected using the ECL system. The reactive bands were quantified using video densitometry and the Biolight for Microsoft Windows, version 2000.01, computer software (Bio-Profil, Vilber Lourmat, France).
Transfection and luciferase reporter assays
The cells were transfected with PG13-luc and Renilla-luc plasmids using the Turbofect reagent. The treated or untreated cells were harvested and the luciferase activity was determined using the Dual-Glo luciferase assay system kit. The luciferase activity was normalized based on the Renilla luciferase activity. The level of luciferase activity was quantified as the ratio of the activity of cells treated with andrographolide to that of untreated control cells.
Suppression of pp2a or shp-1 expression
The suppression of gene was examined as described previously19. For pp2a suppression, predesigned siRNAs targeting the rat pp2a gene were purchased from Ambion (Austin, TX, USA). The sequence of the siRNA oligonucleotide that targeted the coding region of the rat PP2A catalytic subunit (PP2A-C) mRNA was 5′-CCAUACUCCGAGGGAAUCATT-3′. For shp-1 suppression, predesigned siRNAs targeting the rat shp-1 gene were purchased from Sigma-Aldrich (St. Louis, MO, USA). The sequence of the siRNA oligonucleotide that targeted the coding region of the rat SHP-1 mRNA was 5′-CAGUUCAUCGAAACAACCATT-3′. The negative control siRNA contained a 19-bp scrambled sequence with 3′dT overhangs and was also purchased from Ambion (Austin, TX, USA).
PP2A and SHP-1 phosphatase assay
We examined PP2A and SHP-1 phosphorylation using the Immunoprecipitation Phosphatase Assay kit (Millipore, Billerica, MA, USA), which quantifies the phosphorylation of a phosphopeptide substrate. For the PP2A phosphatase assay, 100 μg of cellular proteins was immunoprecipitated with a mouse anti-PP2A-C antibody (1 μg/μl; Millipore). The immunoprecipitated proteins were incubated with 750 μM phosphopeptide (amino acid sequence KRpTIRR) in protein phosphatase assay buffer for 10 min at 30°C to initiate the dephosphorylation of the phosphopeptide substrate. For the SHP-1 phosphatase assay, 100 μg of cellular proteins was immunoprecipitated with a rabbit anti-SHP-1 antibody (0.2 μg/μl; Santa Cruz Biotechnology). The immunoprecipitated proteins were incubated with 300 μM phosphopeptide (amino acid sequence RRLIEDAEpYAARG) in protein phosphatase assay buffer for 10 min at 30°C. The reactions were terminated by the addition of 100 μl of malachite green solution and the absorbance at 650 nm was measured using a microplate reader.
Statistical analysis
The data are expressed as the mean ± standard error of the results and are accompanied by the number of observations. The data were assessed using an analysis of variance. If a significant difference occurred among the group means, each group was compared with the other groups using the Newman-Keuls method. A P < 0.05 was considered to indicate a statistically significant difference.
References
Koh, K. K., Han, S. H., Oh, P. C., Shin, E. K. & Quon, M. J. Combination therapy for treatment or prevention of atherosclerosis: focus on the lipid-RAAS interaction. Atherosclerosis 209, 307–313, 10.1016/j.atherosclerosis.2009.09.007 (2010).
Xu, Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ 10, 400–403, 10.1038/sj.cdd.4401182 (2003).
Sarker, K. P. et al. Ebselen inhibits NO-induced apoptosis of differentiated PC12 cells via inhibition of ASK1-p38 MAPK-p53 and JNK signaling and activation of p44/42 MAPK and Bcl-2. J Neurochem 87, 1345–1353 (2003).
Xiao, W., Ando, T., Wang, H. Y., Kawakami, Y. & Kawakami, T. Lyn- and PLC-beta3-dependent regulation of SHP-1 phosphorylation controls Stat5 activity and myelomonocytic leukemia-like disease. Blood 116, 6003–6013, 10.1182/blood-2010-05-283937 (2010).
Coon, J. T. & Ernst, E. Andrographis paniculata in the treatment of upper respiratory tract infections: a systematic review of safety and efficacy. Planta Med 70, 293–298, 10.1055/s-2004-818938 (2004).
Poolsup, N., Suthisisang, C., Prathanturarug, S., Asawamekin, A. & Chanchareon, U. Andrographis paniculata in the symptomatic treatment of uncomplicated upper respiratory tract infection: systematic review of randomized controlled trials. J Clin Pharm Ther 29, 37–45 (2004).
Burgos, R. A. et al. Efficacy of an Andrographis paniculata composition for the relief of rheumatoid arthritis symptoms: a prospective randomized placebo-controlled trial. Clin Rheumatol 28, 931–946, 10.1007/s10067-009-1180-5 (2009).
Xia, Y. F. et al. Andrographolide attenuates inflammation by inhibition of NF-kappa B activation through covalent modification of reduced cysteine 62 of p50. J Immunol 173, 4207–4217 (2004).
Bao, Z. et al. A novel antiinflammatory role for andrographolide in asthma via inhibition of the nuclear factor-kappaB pathway. Am J Respir Crit Care Med 179, 657–665, 10.1164/rccm.200809-1516OC (2009).
Cheung, H. Y. et al. Andrographolide isolated from Andrographis paniculata induces cell cycle arrest and mitochondrial-mediated apoptosis in human leukemic HL-60 cells. Planta Med 71, 1106–1111, 10.1055/s-2005-873128 (2005).
Li, J., Cheung, H. Y., Zhang, Z., Chan, G. K. & Fong, W. F. Andrographolide induces cell cycle arrest at G2/M phase and cell death in HepG2 cells via alteration of reactive oxygen species. Eur J Pharmacol 568, 31–44, 10.1016/j.ejphar.2007.04.027 (2007).
Zhou, J., Zhang, S., Ong, C. N. & Shen, H. M. Critical role of pro-apoptotic Bcl-2 family members in andrographolide-induced apoptosis in human cancer cells. Biochem Pharmacol 72, 132–144, 10.1016/j.bcp.2006.04.019 (2006).
Cheung, M. T., Ramalingam, R., Lau, K. K., Chiang, W. L. & Chiu, S. K. Cell type-dependent effects of andrographolide on human cancer cell lines. Life Sci 91, 751–760, 10.1016/j.lfs.2012.04.009 (2012).
Zhou, J., Lu, G. D., Ong, C. S., Ong, C. N. & Shen, H. M. Andrographolide sensitizes cancer cells to TRAIL-induced apoptosis via p53-mediated death receptor 4 up-regulation. Mol Cancer Ther 7, 2170–2180, 10.1158/1535-7163.MCT-08-0071 (2008).
Abu-Ghefreh, A. A., Canatan, H. & Ezeamuzie, C. I. In vitro and in vivo anti-inflammatory effects of andrographolide. Int Immunopharmacol 9, 313–318, 10.1016/j.intimp.2008.12.002 (2009).
Hsieh, C. Y. et al. Andrographolide enhances nuclear factor-kappaB subunit p65 Ser536 dephosphorylation through activation of protein phosphatase 2A in vascular smooth muscle cells. J Biol Chem 286, 5942–5955, 10.1074/jbc.M110.123968 (2011).
Hunter, T. Signaling--2000 and beyond. Cell 100, 113–127 (2000).
Hsu, M. J. et al. Apoptosis signal-regulating kinase 1 in peptidoglycan-induced COX-2 expression in macrophages. J Leukoc Biol 87, 1069–1082, 10.1189/jlb.1009668 (2010).
Hsu, M. J. et al. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J Neurosci 27, 5719–5729, 10.1523/JNEUROSCI.1874-06.2007 (2007).
Yin, K. J. et al. Protein phosphatase 2A regulates bim expression via the Akt/FKHRL1 signaling pathway in amyloid-beta peptide-induced cerebrovascular endothelial cell death. J Neurosci 26, 2290–2299, 10.1523/JNEUROSCI.5103-05.2006 (2006).
Yu, Y. et al. GADD45beta mediates p53 protein degradation via Src/PP2A/MDM2 pathway upon arsenite treatment. Cell Death Dis 4, e637, 10.1038/cddis.2013.162 (2013).
Cohen, P. The structure and regulation of protein phosphatases. Annu Rev Biochem 58, 453–508, 10.1146/annurev.bi.58.070189.002321 (1989).
Xie, H. & Clarke, S. Protein phosphatase 2A is reversibly modified by methyl esterification at its C-terminal leucine residue in bovine brain. J Biol Chem 269, 1981–1984 (1994).
Chen, J., Martin, B. L. & Brautigan, D. L. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264 (1992).
Adams, J. M. & Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337, 10.1038/sj.onc.1210220 (2007).
Huang, D. C. & Strasser, A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103, 839–842 (2000).
Hastak, K., Agarwal, M. K., Mukhtar, H. & Agarwal, M. L. Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J 19, 789–791, 10.1096/fj.04-2226fje (2005).
Dumaz, N. & Meek, D. W. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J 18, 7002–7010, 10.1093/emboj/18.24.7002 (1999).
Barr, A. J. et al. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136, 352–363, 10.1016/j.cell.2008.11.038 (2009).
Denicourt, C. & Dowdy, S. F. Medicine. Targeting apoptotic pathways in cancer cells. Science 305, 1411–1413, 10.1126/science.1102974 (2004).
Fulda, S., Galluzzi, L. & Kroemer, G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 9, 447–464, 10.1038/nrd3137 (2010).
Marzo, I. et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–2031 (1998).
Hastak, K., Agarwal, M. K., Mukhtar, H. & Agarwal, M. L. Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J 19, 789–791, 10.1096/fj.04-2226fje (2005).
Jeffers, J. R. et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4, 321–328 (2003).
Chang, H. L. et al. Simvastatin induced HCT116 colorectal cancer cell apoptosis through p38MAPK-p53-survivin signaling cascade. Biochim Biophys Acta 1830, 4053–4064, 10.1016/j.bbagen.2013.04.011 (2013).
el-Deiry, W. S. et al. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993).
Takeda-Matsubara, Y. et al. Estrogen activates phosphatases and antagonizes growth-promoting effect of angiotensin II. Hypertension 39, 41–45 (2002).
Kundu, S. et al. Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J Immunol 184, 6529–6536, 10.4049/jimmunol.0903562 (2010).
Zhang, J., Somani, A. K. & Siminovitch, K. A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol 12, 361–378, 10.1006/smim.2000.0223 (2000).
Neel, B. G., Gu, H. & Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28, 284–293, 10.1016/S0968-0004(03)00091-4 (2003).
Frank, C. et al. Effective dephosphorylation of Src substrates by SHP-1. J Biol Chem 279, 11375–11383, 10.1074/jbc.M309096200 (2004).
Lu, Y. et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem 278, 40057–40066, 10.1074/jbc.M303621200 (2003).
Chen, Y. Y., Hsu, M. J., Sheu, J. R., Lee, L. W. & Hsieh, C. Y. Andrographolide, a Novel NF- kappa B Inhibitor, Induces Vascular Smooth Muscle Cell Apoptosis via a Ceramide-p47phox-ROS Signaling Cascade. Evid Based Complement Alternat Med 2013, 821813, 10.1155/2013/821813 (2013).
Bao, G. Q. et al. Andrographolide causes apoptosis via inactivation of STAT3 and Akt and potentiates antitumor activity of gemcitabine in pancreatic cancer. Toxicol Lett 222, 23–35, 10.1016/j.toxlet.2013.06.241 (2013).
Goldman, E. H., Chen, L. & Fu, H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3-3 dissociation. J Biol Chem 279, 10442–10449, 10.1074/jbc.M311129200 (2004).
Wei, M. C. et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730, 10.1126/science.1059108 (2001).
Yang, S. et al. Mitochondrial-mediated apoptosis in lymphoma cells by the diterpenoid lactone andrographolide, the active component of Andrographis paniculata. Clin Cancer Res 16, 4755–4768, 10.1158/1078-0432.CCR-10-0883 (2010).
Zhang, H. M. et al. Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol Cell Biol 25, 6247–6258, 10.1128/MCB.25.14.6247-6258.2005 (2005).
Hsu, Y. F. et al. p53 in trichostatin A induced C6 glioma cell death. Biochim Biophys Acta 1810, 504–513, 10.1016/j.bbagen.2011.02.006 (2011).
Baeuerle, P. A. & Baltimore, D. NF-kappa B: ten years after. Cell 87, 13–20 (1996).
Hsiao, G. et al. A novel antioxidant, octyl caffeate, suppression of LPS/IFN-gamma-induced inducible nitric oxide synthase gene expression in rat aortic smooth muscle cells. Biochem Pharmacol 65, 1383–1392 (2003).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65, 55–63 (1983).
Asanuma, K., Campbell, K. N., Kim, K., Faul, C. & Mundel, P. Nuclear relocation of the nephrin and CD2AP-binding protein dendrin promotes apoptosis of podocytes. Proc Natl Acad Sci U S A 104, 10134–10139, 10.1073/pnas.0700917104 (2007).
Lin, K. H., Hsiao, G., Shih, C. M., Chou, D. S. & Sheu, J. R. Mechanisms of resveratrol-induced platelet apoptosis. Cardiovasc Res 83, 575–585, 10.1093/cvr/cvp139 (2009).
Acknowledgements
This work was supported by grants from the National Science Council, Taiwan (NSC97-2320-B-038-016-MY3, NSC98-2320-B-038-007-MY3, NSC100-2320-B-038-021-MY3 and NSC101-2320-B-038-034), Shin Kong Wu Ho-Su Memorial Hospital (SKH-8302-102-NDR-04).
Author information
Authors and Affiliations
Contributions
Y.Y.C. and M.J.H. contributed to the research design, performed the research, analyzed the results and wrote the draft of the manuscript. C.Y.H., K.H.L. and W.J.L. performed some experiments. D.S.C. and T.J. provided intellectual input and critically reviewed the manuscript. M.J.H. and J.R.S. conceived and supervised the study, analyzed the results and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Figures with title page
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chen, YY., Hsieh, CY., Jayakumar, T. et al. Andrographolide induces vascular smooth muscle cell apoptosis through a SHP-1-PP2A-p38MAPK-p53 cascade. Sci Rep 4, 5651 (2014). https://doi.org/10.1038/srep05651
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05651
This article is cited by
-
Mitochondrial apoptosis is induced by Alkoxy phenyl-1-propanone derivatives through PP2A-mediated dephosphorylation of Bad and Foxo3A in CLL
Leukemia (2019)
-
1 billion-year-old cell contents preserved in monazite and xenotime
Scientific Reports (2019)
-
PGA2 induces the expression of HO-1 by activating p53 in HCT116 cells
Molecular & Cellular Toxicology (2017)
-
Accumulation of p53 via down-regulation of UBE2D family genes is a critical pathway for cadmium-induced renal toxicity
Scientific Reports (2016)
-
Andrographis paniculata transcriptome provides molecular insights into tissue-specific accumulation of medicinal diterpenes
BMC Genomics (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
