
Abstract
The field of single-cell analysis has gained a significant momentum over the last decade. Separation and isolation of individual cells is an indispensable step in almost all currently available single-cell analysis technologies. However, stress levels introduced by such manipulations remain largely unstudied. We present a method for minimally invasive retrieval of selected individual adherent cells of different types from cell cultures. The method is based on a combination of mechanical (shear flow) force and biochemical (trypsin digestion) treatment. We quantified alterations in the transcription levels of stress response genes in individual cells exposed to varying levels of shear flow and trypsinization. We report optimal temperature, RNA preservation reagents, shear force and trypsinization conditions necessary to minimize changes in the stress-related gene expression levels. The method and experimental findings are broadly applicable and can be used by a broad research community working in the field of single cell analysis.
Similar content being viewed by others
Introduction
The field of single cell analysis has experienced a tremendous growth over the last decade owing to the intense interest in intercellular heterogeneity and its functional role at the tissue level and disease states in vivo1,2,3,4. New technological advancements have enabled the exploration of biological phenomena with single-cell resolution5,6,7,8. Almost all existing methods for single-cell analysis that require isolation of individual cells involve some type of mechanical transportation or manipulation of single cells for sample preparation and/or analysis purposes. Current technologies for retrieving single cells from cell culture include micromanipulation6,8,9, laser capture microdissection10 and microfluidics11. One of the current technological challenges is the minimization of perturbation to the cells as a result of such transportation to make biologically relevant inferences about cell function possible. If the resulting stress to the cell is significant it can alter cellular profiles at the physiological, gene transcription and/or expression levels and confound experimental results. Although widely used, stress levels introduced to cells by manipulation and, more importantly, their potential effects on cell function remain largely unknown. Mechanical cues and mechanical stress have been found to strongly affect most cellular functions and critically influence gene transcription during embryogenesis, organogenesis12 and embryonic vasculature development13. Mechanical stress also exhibits a direct effect on the nuclear architecture-mediated gene transcription regulation14, oncogenesis15, stem cell differentiation, cancer metastasis and the immune response16 among others. It is thus likely that mechanical stress introduced during cell manipulation can significantly alter gene expression in cells resulting in atypical both gene expression profile and cellular function. Therefore, characterization of stress levels that can significantly perturb cell function is necessary for studies that utilize single-cell analysis techniques.
In the context of single-cell analysis methods, perturbations can be divided into two major categories with regard to time scales. One category is perturbations that cause reversible alterations that occur on a timescale that is much shorter than the time between the perturbation and analysis. By definition, perturbations of this type do not result in significant changes in the cell at the time of analysis and thus can be considered negligible. The second category is perturbations that induce a long-lasting (on timescales comparable or longer than the time between stress administration and analysis) response in the form of a modified gene expression profile. These perturbations can introduce modifications to the cell function, mRNA or protein expression levels or all of them simultaneously and thus need to be properly assessed before reaching any conclusions about experimental findings. It is likely that adherent cell types should be affected by manipulation more than non-adherent cells simply due to the fact that the former need to be detached from the growth substrate or dissociated from tissue before any kind of manipulation can be performed. Owing to changes in cellular tension, the detachment step itself could cause the cell to respond with an altered gene expression profile mediated by mechanosensing through e.g. integrin-actin linkages and mechanostransduction via downstream signaling cascades such as receptor-type tyrosine-protein phosphatase alpha (RPTP-α), Src family kinases (SFKs)17,18,19, focal adhesion kinase (FAK)20,21 and others. In addition, any type of manipulation can induce additional cellular responses at biomolecular and/or organelle levels. Epithelial cells adhere to the extracellular matrix through transmembrane adhesion protein complexes. At the basal membrane, the adhesion of epithelial cells to the extracellular matrix is built upon different types of cell-ECM adhesions, including focal adhesions and hemidesmosomes, both of which are mediated by integrin connections22, nascent adhesions, focal complexes, focal adhesions, podosomes and others23. These protein complexes, including integrin-actin networks and integrin-intermediate filament networks, regulate the adhesion but also mediate mechanosensing and signal mechanotransduction into the cell24. To remove cells from a given culture substrate, various mechanical and chemical methods have been employed. For instance, proteolytic enzymes, such as trypsin, or chelators, can break the integrin-ligand bonds that mediate cell attachment to the substrate25. However, enzymatic dissociation can damage cells, especially the cell surface. Moreover, alterations of gene expression levels in cells treated with trypsin were discovered using global gene expression profiling on the microarray platform26. Therefore, trypsinization should be performed with caution, by optimizing both the duration of trypsinization and the concentration of trypsin.
Mechanical means such as scraping or shear flow were employed to remove cells from substrates27. However, mechanical methods are usually disruptive to the cells and potentially result in a loss of cellular contents. Shear flow is less disruptive than scraping because the latter tends to damage the plasma membrane. Still, even under relatively mild conditions, shear stress can affect signal transduction pathways, especially in endothelial cells28. Because epithelial cells experience much less shear stress in the body, gene expression alterations within those cells in response to shear stress could be significant and need to be explored. So far, expression levels of chemokine (C-C motif) ligand 2 (CCL2) have been shown to be upregulated in epithelial cells in response to shear flow stress29. The use of mechanical force or biochemical treatment alone is more likely to perturb gene expression when retrieving single cells. It is however likely that, when biochemical treatment and shear flow are combined, a relatively mild treatment can remove cells from the surface and transfer them without causing damage to the plasma membrane and alterations in gene expression levels. Therefore, an optimal range of shear flow force and trypsinization conditions for retrieving individual cells needs to be determined for minimizing alterations of the gene expression profile.
In this study we developed a new method for retrieval of individual adherent cells that is based on a combination of mechanical forces and biochemical treatment. Our goal was to develop a single-cell retrieval technique that enables unbiased measurements of gene expression levels in individual cells right after the retrieval. To achieve this goal we investigated stress response induced by the method in terms of alterations at the gene expression level in individual cells. To account for differences in adhesion strength among different cell types, we used three types of epithelial cells with different adhesion to substrate strengths. We quantified alterations in expression levels of stress response-related genes in single cells as a function of varying mechanical stress while employing both shear flow and trypsin digestion to detach single cells from glass substrates. We optimized the treatment conditions in a highly controllable manner to minimize the effects of gene expression changes that could be induced by stresses. We report an optimal range for mechanical force needed to efficiently detach single cells with no detectable change in expression levels of the studied stress-response genes. We expanded the utility of this technique to distinguish and harvest co-cultured cells from microwells using a fluorescence-assisted single-cell harvesting method. These findings can be useful for studies focused on single-cell analysis that involve any mechanical manipulation of live cells. Because the technique does not induce measurable changes in gene expression levels in the short term, it is reasonable to expect that long-term effects on cell function will also be minimal or absent. This further extends the utility of the technique to a broad variety of applications that require a re-culturing of the retrieved individual cells e.g. for clonal expansion.
Results and Discussion
Comparison between in-situ direct lysis and “pick-and-place” harvesting of single cells in microwells
In order to perform end-point gene transcription analysis of single cells, the total RNA from individual cells needs to be harvested with minimal loss. We first tested the ability of different single-cell harvesting methods to preserve and recover the maximum amount of total RNA using several different buffers. The in-situ direct lysis of single cells in microwells (Figure 1A) contained three steps: (1) lysing single cells in microwells; (2) aspirating the lysate using a micropipette and a custom high-precision pump; (3) dispensing the lysate into the cap of a microcentrifuge tube for RT-qPCR analysis. The in-situ direct lysis method was initially used to eliminate the steps of cell detaching and transferring from microwells to analysis vials and avoid potential stress during the cell detachment process. As a comparison to the first method, a “pick-and-place” cell harvesting method7,30 using the single-cell manipulation platform8 developed in our lab was tested (Figure 1B, Figure S1). The method combines trypsinization and shear flow to detach single cells from the bottom of the microwells and collect them for downstream analysis, e.g. RT-qPCR (Figure 1C–E). For the study we used three different human esophageal epithelial cell lines derived from normal, metaplastic and late dysplastic regions in the esophagus. The choice of the cell types was made to demonstrate the applicability of the method to cells with varying adhesion to substrate strengths. Metaplastic (CP-A cell line) cells exhibit highest adhesion strength as compared to normal (EPC-2) or dysplastic (CP-D) cells. CP-A cells in microwells were treated with 0.05% Trypsin for 6 minutes until they were partially detached as judged from the change in the cellular morphology from fully stretched to a more spherical shape. Single cells were then collected from the microwells using the micropipette in the single-cell manipulation platform using a shear force of 4.5 nN.

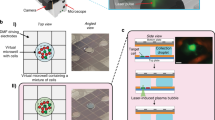
The single-cell harvesting platform design and schematic view of harvesting steps.
(A) Microwell array design. 3 × 3 arrays of wells with 300-μm center-to-center spacing were fabricated on fused silica wafers using hydrofluoric acid (HF) deep wet etch lithography. Each well is 20-μm deep and has a diameter of 50 μm. (B) The single-cell manipulation platform uses a micropipette controlled by a piezoelectric pump. Single cells can grow in microwells which are glued to the bottom of the Petri dish. The micropipette is used for aspirating and dispensing single cells. The single-cell harvesting procedure contains three major steps: (C) trypsinization to partially detach the cell from the substrate; (D) trypsin deactivation with trypsin inhibitor (DTI) and cell aspiration into the micropipette tip; (E) transfer of the cell into the cap of a PCR tube for downstream analysis.
Using the in-situ direct lysis method, the single cells were immediately lysed in microwells by adding the RNA lysis buffer into the microwell. The addition of the lysis buffer resulted in substantial morphological changes. We observed that the lysate diffused out of the microwell prior to being collected rendering this method inappropriate for collecting total RNA from single cells. Compared with in-situ direct lysis, single-cells can be harvested without physical tearing or morphological changes to the cell using the pick-and-place harvesting method. Therefore, the pick-and-place harvesting was further developed and optimized in later experiments.
Use of cellular RNA preservation solution for single-cell harvesting
Stabilization of the total RNA is an important aspect of gene transcription assays. To preserve the RNA quality, RNA degradation by RNase should be minimized during cell harvesting and until RNA extraction. Three different solutions - RNA Later®, RNA Lysis Buffer and Keratinocyte serum-free medium - were tested for their ability to preserve RNA in single cells harvested at room temperature. We measured expression levels of the 28S and actin β (ACTB) housekeeping genes. Both genes are highly expressed in human cells and show low cell-to-cell copy number variability. They can easily be detected using qPCR and used as references for total RNA extraction efficiency in comparison studies. Compared with the quantification cycle (Cq) values of the two genes in the group where only cell medium was used for storing the harvested cells, the Cq values of the two genes in both the RNA Later® and the Lysis buffer groups are higher. The differences in Cq values of 28S and ACTB genes are shown in Figure 2. Cq of both genes in the lysis buffer groups are significantly different from that of the medium group (p = 0.02 for 28S, p = 0.04 for ACTB, calculated using the non-parametric Mann-Whitney statistical significance test, α = 0.05, two-tailed). Higher amounts of 28S and ACTB mRNA in the medium group indicate that the medium outperforms RNA Later® and lysis buffer in preserving RNA. RNA Later® contains ammonium sulfate which can permeate the cellular membrane and lead to a leakage of cellular components including RNA31. In addition, we were not able to spin down pico-gram levels of single cell RNA from RNA Later® solution, which may have further reduced the RNA extraction efficiency. RNA lysis buffer, on the other hand, lyses cells almost instantaneously, potentially exposing the picogram levels of single cell RNA to environmental RNases. We also tested another RNA-preserving agent, RNAstable (Biomatrica, San Diego, CA, USA) by adding 200 μL of RNAstable LD into the cap prior to harvesting. Results (data not shown) indicated that RNAstable LD is inferior in preserving cellular RNA as compared with the medium. Based on these results we find that the Keratinocyte cell growth medium exhibits the highest level of protection against RNA degradation/loss among the four solutions. Based on this finding, the single-cell collection method was optimized by using 200 μL of Keratinocyte SFM in the 1.5-mL microcentrifuge tubes.

Comparison of RNA preservation levels between medium, RNALater and RNA Lysis Buffer during single-cell harvesting.
The quantification cycle (Cq) values are higher in the lysis buffer group for the 28S (Panel A) and higher in the RNALater group for the ACTB gene (Panel B) as compared with the medium group. The corresponding Cq mean values are shown on top of the bars to compare the three conditions. Errors and error bars are corresponding standard deviations. The number of cells analyzed (n) for each harvesting condition is shown at the bottom of the graphs. Each qPCR reaction was run with three technical replicates. The difference between the lysis buffer and medium preservation group is statistically significant for the 28S gene and between the RNALater and medium group for the ACTB gene (both tested with the two-tailed Mann-Whitney test, α = 0.05). Therefore, dispensing cells in cell culture medium can better preserve mRNA.
Low temperature harvesting for preserving cellular RNA
Low temperatures are known to inhibit enzymatic activity of cellular proteins, including RNase. To determine whether lowering the temperature can facilitate RNA preservation due to RNase inactivation, two ice packs were placed on the stage of the single-cell manipulation platform for 15 min before the harvesting experiment to pre-cool the platform. During the entire harvesting procedure, the Petri dish containing cells in the microwells was surrounded by the ice packs. Room temperature harvesting without ice pack cooling was used as the control. RT-qPCR analysis was performed on three genes-28S, ACTB and HSP70 from the harvested cells to assess the preservation efficiency. The HSP70 gene was chosen due to its role in cold response32 and cellular stress response in general33,34. Gene transcription levels of all three genes in the ice cooled harvesting group do not exhibit statistically significant changes compared with room temperature harvesting control (Figure 3). This indicates that the mRNA of the studied genes is stable under both cool and room temperature conditions, while other genes, such as ACTB, may be more sensitive to degradation at room temperature. Furthermore, the unchanged level of HSP70 mRNA indicated that the low temperature condition did not introduce an additional stress factor to the cells. Even though we did not observe statistically significant differences between the room temperature and the ice-cooled conditions for the studied genes, we used the ice-cooled condition throughout the study to avoid potential degradation of the total RNA at room temperature.

Comparison between low temperature and room temperature harvesting conditions for preserving cellular RNA.
Three individual cells were harvested for each condition and each qPCR reaction was run with three technical replicates. The differences between the Cq mean values (shown above each box with S.D. as error) between the two groups in the 28S, ACTB and HSP70 genes are not statistically significant (Mann-Whitney test, α = 0.05, two-tailed). Error bars represent the standard deviations.
The effects of flow-rate and trypsinization time on harvesting success rate and RNA preservation
In order to detach adherent cells from the microwell glass surface with minimal perturbation, a combination of mechanical force and enzymatic digestion was used. Shear flow through the micropipette tip can aspirate single cells out of the microwell; however, applying shear flow can result in damages to the plasma membrane and loss of the cellular RNA. Trypsin is a traditional protease which can cleave membrane adhesion proteins, primarily integrins and detach the cells from the substrate. Excessive trypsinization, however, especially with regard to trypsinization time, can alter the gene expression profile and affect cell viability35,36. To minimize potential perturbations to the gene transcription profile and the loss of total RNA during harvesting, we have employed a combination of both methods. We first performed trypsinization which was followed by applying a shear flow to harvest single cells. Because either step can damage the cell integrity and/or mRNA profile, we have optimized the approach by testing different combinations of mechanical, chemical and temporal parameters. Our purpose was to examine how the trypsinization time, trypsin concentration and shear flow affect the harvesting success rate (HSR) and gene transcription levels. The main goal was to identify the lowest flow-rate and shortest trypsinization time needed to achieve reliable detachment of cells with the highest HSR while causing minimal changes in gene transcription levels.
A total of nine different conditions (Table 1) were tested, including three shear forces (0.9 nN, 4.5 nN, 9.0 nN) and three trypsinization times (5 min, 8 min, 10 min).
Fifteen cells were treated under each condition and the successfully harvested cells were used for RT-qPCR analysis (Table S1). The success rate of cells harvested under each condition without taking into account cell stress levels demonstrates that trypsinizing cells for 10 minutes and harvesting at a shear force of 9.0 nN has the highest success rate of 93.3% (Table 1).
To assess the stress levels resulting from the different harvesting conditions, we analyzed the mRNA levels of 28S, ACTB, HSP70 and CCL2 genes, in harvested cells using RT-qPCR. We pooled the cDNA extracted from single cells obtained for each harvesting condition and performed qPCR on them. The results show that the transcription levels of the HSP70, 28S and CCL2 genes are generally higher in cells harvested after 5 min trypsinization as compared to cells harvested after 8 or 10 min of trypsinization. The difference in their expression levels in cells between 5 min, 8 min and 10 min trypsinization groups is statistically significant (Table S1 and Table S2). Moreover, the transcription levels of the ACTB, 28S and CCL2 genes are markedly affected by the shear force. In general, the shear force of 4.5 nN yields the lowest Cq values for these genes. Different combinations of the shear force and trypsinization time also significantly affect the gene transcription levels of the ACTB, 28S and CCL2 genes. The two-way multivariate ANOVA test (α = 0.05) performed on the Cq values as a function of trypsinization time (TT) or shear force (SF) shows (Table 2) that both the trypsinization time and shear force have a statistically significant impact on the transcription levels of these genes. If the cells are trypsinized for a shorter time, a higher shear force needs to be applied to retrieve the cells and vice versa. Interestingly, the p-values of the two factors (Table 2, combined SF and TT p-values) indicate that enzymatic cleavage and mechanical shear flow have a collective effect on the transcription of the ACTB, 28S and CCL2 genes. Since shorter trypsinization time and low shear force are desired for preserving cellular RNA, we limited the trypsinization time to 5–7 minutes and utilized a shear force of 4.5 nN for single cell harvesting.
Comparison between on-chip direct lysis of cells in microwells and harvesting cells from microwells
With the preservation medium, temperature, trypsinization time and flow-rate conditions optimized, we wanted to determine whether the harvesting procedure itself under those conditions causes any observable RNA loss or changes in gene transcription. To this end we compared the amounts of mRNA extracted from cells that were harvested with those that did not undergo harvesting. We directly lysed individual cells in microwells on the chip. Because the direct lysis step does not require trypsinization or shear flow, we use this condition as the untreated control group. If no significant differences in gene transcription levels of the selected genes can be detected between the control group and the harvested group, the harvesting procedure can be considered suitable to collect cells for gene transcription analysis.
In the on-chip direct lysis experiment, 9 single cells were loaded into a 3 × 3 array of microwells made in fused silica chips and the chips were placed in a Petri dish. The cells on the chip were incubated overnight. After being visually inspected under the microscope for cell occupancy in the microwells, the glass chips with cells adhered to the well bottom were placed into a 1.5-mL microcentrifuge tube containing 200 μL of RNA Lysis Buffer.
In the harvesting experiment, single-cells were harvested from the microwells on the cooled stage, using a shear force of 9.0 nN and 6 min trypsinization time and placed into the cap of a microcentrifuge tube containing 200 μL of cell growth medium. The mRNA levels of the 28S, ACTB, GAPDH and HSP70 genes in cells directly lysed on the chip or harvested were analyzed using RT-qPCR.
Cq values of the four genes were compared between the on-chip direct lysis and harvesting groups to assess the capability of the two methods to preserve cellular RNA. The Cq values of 28S, ACTB, GAPDH and HSP70 genes in harvested cells were found to be very close to those measured in on-chip directly lysed cells (Figure 4A).The Mann-Whitney test showed that the Cq differences (ΔCq = Cq, harvest − Cq, direct lysis) between the two groups in all of the genes are not statistically significant (Figure 4B–E). This suggests that the parameters used for harvesting preserve cellular RNA efficiently and do not induce detectable changes in the mRNA profile or in the amount of the total RNA extracted from the cell.

Comparison of gene transcription levels between the direct lysis of cells in microwells and harvesting cells from microwells.
Each 3 × 3 microwell array was analyzed using qPCR with three technical replicates for each gene. (A) Cq values of the 28S, ACTB, GAPDH and HSP70 genes obtained with the harvested cells are very close to those measured with cells lysed directly in the microwells (no harvesting). The corresponding Cq mean values are shown on top of the bars. Error bars show the standard deviations. ΔCq between the two groups in all of the genes (Panel B–E) are not statistically significant (Mann-Whitney test, n = 6, α = 0.05, two-tailed).
Fluorescence-assisted single cell harvesting
Cell-cell communications are critical to regulating various core cellular responses, such as metabolism and homeostasis. Understanding gene transcription changes as a result of intercellular interactions in cells at different stages of pre-malignant progression may help discover new cancer biosignatures. To enable harvesting of individual cells of different types from co-cultured cell populations, we added fluorescence-assisted single-cell harvesting modality to the method. To perform cell harvesting with minimal effect on gene transcription levels we used harvesting parameters optimized in the cell stress as presented above. Using this approach, we can distinguish co-cultured cells of different types utilizing cell-type specific fluorescent markers and separately collect individual cells from a single microwell.
To this end we produced two cell lines each expressing a different fluorescent protein. Normal epithelial EPC-2 cells were transfected with Lentiviral vectors expressing cytosolic FP635 to establish the EPC-2/FP635 cell line. Dysplastic Barrett's Esophagus CP-D cells were transfected with Lentiviral vectors expressing cytosolic TurboGFP to establish the CP-D/TurboGFP cell line. Co-cultured EPC-2/FP635 and CP-D/TurboGFP cells could easily be distinguished under the fluorescence microscope (Figure 5 A–D). The single-cell manipulation platform7,8 was equipped with an epi-fluorescence illumination source and excitation/emission filter sets for TurboGFP and FP635. With the help of a custom-written program (LabView, National Instruments, Austin, TX) for fluorescence-assisted cell harvesting, EPC-2/FP635 cells and CP-D/TurboGFP cells can be identified within the co-culture in the microwells. They can be detached and collected separately by the micropipette using the optimized harvesting conditions.

Harvesting single cells from a co-culture.
One CP-D/TurboGFP cell and One EPC-2/FP635 cell were co-incubated in microwells. Micrographs of: (A) bright field; (B) overlay of green and red channel with bright field channel; (C) CP-D/TurboGFP cell in green spectral channel, (D) EPC-2/FP635 in red spectral channel. Co-cultured EPC-2/FP635 and CP-D/TurboGFP cells can be distinguished and harvested using the fluorescence-assisted single cell harvesting platform.
In order to demonstrate the ability of our technique to distinguish specific signatures of single cells, we measured the presence of TurboGFP transcripts in harvested cells using RT-qPCR. We expected that the Cq values of the Turbo-GFP gene in CP-D/TurboGFP cells are significantly lower compared with those in EPC-2/FP635 cells. The amplification plots of RT-qPCR showed the presence of TurboGFP in CP-D/TurboGFP cells and marked differences in Cq in EPC-2/FP635 cells (Figure 6A). Normalized Cq values using CP-D/TurboGFP cells as the reference demonstrated a significant difference in signal between two types of cells as determined by the Wilcoxon test (α = 0.05, two-tailed) with p = 0.0009 (Figure 6B).

RT-qPCR analysis of TurboGFP genes in CPD/TurboGFP and EPC2/FP635 cells.
Different cell types can be reliably distinguished using the fluorescent assisted harvesting method as proven by the RT-qPCR results. Quantitative cycle values on the amplification plots (A) and ΔCq analysis using CPD/TurboGFP cells as the control (B) both demonstrate the presence of TurboGFP in harvested CPD/TurboGFP cells but not in EPC2/FP635 cells.
Conclusions
In the study, we developed a method for retrieving adherent cells from substrates with minimal disruption and perturbation. The method features combined enzymatic cleavage and mechanical force applied by a piezo-pump on a single cell manipulation station. Reagents for RNA preservation, temperature settings, shear force and trypsinization time were also optimized to minimize gene transcription profile changes brought by harvesting. Using this method, gene transcription levels were analyzed in both harvested and on-chip directly-lysed single cells. The results showed conclusively that the harvesting method we developed and optimized can preserve the RNA profiles in the cells retrieved from microwells. Among the three types of cells we harvested, the metaplastic epithelial cells (the CP-A cell line) showed the highest adhesion to substrate strength and needed the longest trypsinization time – 8–10 minutes - for optimal retrieval. CP-D (dysplastic) and EPC-2 (normal) epithelial cells exhibited lower adhesion to substrate strength compared to CP-A cells, needed shorter trypsinization times (5–7 minutes) and were easier to retrieve than CP-A cells when using same shear force. We expanded the application to fluorescence-assisted single cell harvesting from a co-culture of different cell types. This method provides an approach to transfer adherent single cells from cell culture to any downstream end-point analysis. We note that to extract individual cells of other cell types from various structures, including tissues and 3D culture matrices, it would be critical to find an optimal range for the enzymatic treatment and shear force that would enable minimally invasive retrieval of individual cells while keeping the structure intact. The method enables researchers to retrieve adherent single cells without perturbation and thus has the potential to become a broadly applicable tool in the growing field of single-cell analysis.
Methods
Microwell design
Arrays of 3 × 3 microwells were used for culturing single-cells. Each microwell has an inner diameter of 50 μm and is 20 μm deep (Figure 1A)37. The microwells were fabricated in fused silica substrates using wet-etch lithography. The dimensions of these microwells were optimized for cell-cell interaction studies to monitor cellular gene expressions in a controlled microenvironment.
Cell culture
The normal cell line, EPC-238, the metaplastic cell line, CP-A and the pre-malignant cell line, CP-D, were derived from healthy, metaplastic and dysplastic human esophageal regions in Barrett's Esophagus39, respectively. Cells were cultured using Gibco keratinocyte serum-free cell growth medium (Invitrogen, Carlsbad, CA, USA), supplemented with hEGF (Peprotech, Rocky Hill, NJ, USA) at 2.5 μg/500 mL, BPE (bovine pituitary extract) at 25 mg/500 mL and penicillin–streptomycin solution (Invitrogen) at 100 units/100 μg/mL. Cells were grown at 37°C under 5% CO2. Prior to experimentation, cells were cultured in a 75 cm2 flask to approximately 80% confluency.
Cell loading into microwells
Individual cells were loaded into microwells using a single-cell manipulation platform8. Briefly, the platform is built around a diaphragm micropump that can aspirate and dispense sub-nanoliter volumes of liquid. Single cells in suspension were aspirated and dispensed into microwells using a 40-μm diameter glass capillary micropipette utilizing closed-loop microscopic vision-based feedback. Cells can be aspirated from a Petri dish by a drag force generated through a negative pressure applied to the micropipette capillary. Cells can be dispensed through the micropipette capillary into the microwell by applying a positive pressure to the capillary, generating an ejection force on the cell7. Glass substrates containing 3 × 3 arrays of microwells were glued to the bottom of a Petri dish with a pre-cut hole using medical-grade epoxy glue (K45-S-14ML, Chemical Concepts, Huntingdon Valley, PA). Loading 9 single-cells in a 3 × 3 array of microwells with one cell per well takes approximately 5–8 minutes, while loading two cells per well requires 20–25 minutes. After loading into the microwells, the cells were incubated in Keratinocyte SFM, at 37°C under 5% CO2 for 16–24 hours to allow for cell adhesion and recuperation from potential stress caused by manipulation.
Direct lysis of single cells in microwells
The microwell substrate was placed on the stage of a pick-and-place single-cell manipulation platform. The micropipette was filled with Cell Lysis Buffer (Zymo Research, Orange, CA, USA) and lowered until it touched the substrate. The micropipette orifice was aligned with a selected cell inside a microwell and encapsulated by the micropipette tip. The Cell Lysis Buffer was dispensed for 1 minute with enough volume to thoroughly coat the cell. After this, the lysate was immediately aspirated into the micropipette for 1 minute to minimize the diffusion of the lysate from the microwell. The lysate was then dispensed through the micropipette into the cap of a 1.5 mL microcentrifuge tube for 1 minute.
Single-cell collection
The pick-and-place single-cell manipulation platform7,8 (Figure S1) was used to collect single-cells from microwells. The microwell substrate containing individual cells was washed three times with 1 mL of warmed 1× PBS and exposed to 1 mL of 0.05% v/v trypsin-EDTA for 5–10 minutes at 37°C. Subsequently, 1 mL of trypsin inhibitor (DTI) was added to the trypsinized cells to deactivate trypsin, which was followed by adding 1 mL of Keratinocyte SFM medium. The selected cell was first aligned with the micropipette orifice and then aspirated into the micropipette capillary. The aspirated cell was dispensed from the microcapillary tip into the cap of a 1.5-mL microcentrifuge tube (VWR, West Chester, PA, USA) containing 200 μL of Keratinocyte SFM, RNA Lysis Buffer, or RNAlater (Life Technologies, Austin, TX, USA). The RNA Later® solution (Ambion, Austin, TX, USA) is a concentrated salt solution (25 mM Sodium Citrate, 10 mM EDTA, 70 g ammonium sulfate/100 ml solution, pH 5.2) that rapidly permeates tissues to stabilize and protect cellular RNA40. RNA Later® was mixed with Keratinocyte serum-free medium at a 5:1 v/v ratio in our experiments. RNA Lysis Buffer (Zymo Research, Orange, CA, USA) is composed of guanidinium thiocyanate, which can both lyse cells and deactivate RNases by denaturing them41. Keratinocyte serum-free medium was used as a control for RNA preservation experiments. The tube was immediately closed and placed on dry ice.
Preservation of RNA at low temperatures
After trypsin deactivation, the substrate containing the 3 × 3 array of microwells with cells was placed inside a 4°C refrigerator for 15 minutes. Simultaneously, the pick-and-place manipulation platform was cooled using four ice-packs for 15 minutes. The substrate was placed on the cooled stage and the temperature was monitored with a thermometer, ranging from 2°C to 10°C on the station. The pick-and-place cell manipulation system was used to aspirate the harvested cells and dispense them into caps of 1.5-mL microcentrifuge tubes as described in the Single-cell collection section above. A control group of cells was kept at room temperature (25°C) during the harvesting process.
Shear force and trypsinization time
The two major determinants of successful cell detachment are the trypsinization time and the applied shear force. Each trypsinization time point and shear force was selected based on preliminary experiments that showed a range of possible values that demonstrated noticeable changes in detachment, from no detachment, partial detachment to full detachment. Shear force was calculated based on numeric simulations of the flow rate (Supplementary Materials). The latter was calculated from the voltage change rate of the piezoelectric pump to volume change rate (Figure S2, Table S2). To measure the success of each combination of shear flow and trypsinization time (Table 1), the micropipette tip was positioned vertically at a 90 degree angle with the substrate surface so that it enclosed the cell without making direct contact with the cell. A selected shear flow was then applied to aspirate the cell from the substrate. A total of fifteen cells were tested for each harvesting condition. A successfully harvested cell is considered one that detaches without visible tearing of the plasma membrane and leaving remnants on the substrate. After a visual inspection and confirmation, the harvested cells were dispensed through the micropipette capillary into the caps of 1.5-mL microcentrifuge tubes as described in the Single-cell collection section above. The harvesting success rate (HSR), the number of cells successfully harvested divided by total number of cells tested (n = 15), is summarized in Table 1.
On-chip lysis of single cells in microwells
For on-chip lysis of single cells, individual cells were loaded into microwells using the pick-and-place single-cell manipulation platform7,8. Glass substrates containing 3 × 3 arrays of microwells were simply placed in a Petri dish, rather than glued to the bottom of the dish. A total of 9 cells were loaded into the 3 × 3 array of microwells (1 cell/well). The cells were incubated in Keratinocyte SFM, at 37°C under 5% CO2 for 16–24 hours. The cells in the microwells were visually inspected under the microscope prior to the experiment for occupancy and/or morphological abnormalities. After incubation we observed an average of 7 out of 9 cells in the microwells due to cell motility, with the remaining 2 cells usually located in the interstitial area just outside of the microwells. The microwell chips were picked up and placed into a 1.5-mL Eppendorf tube containing 200 μL of RNA Lysis Buffer (Zymo Research, Orange, CA, USA) using tweezers. The cells on the chip were lysed in the tube for 30 seconds. After that, the microwell chip was taken out of the tube and RNA isolation from the lysis buffer was immediately performed.
RNA isolation, reverse transcription and qPCR
Single cell RNA isolation, reverse transcription and qPCR were performed as previously described30,42. A description of the protocol is provided in the Supplementary materials and methods. The nonparametric Mann–Whitney test was used for statistical significance analysis.
Lentiviral transfection of cells
The CP-D and EPC-2 cell lines were tagged with TurboGFP and TagFP635 through lentiviral transfection, respectively, to distinguish different cell types in one microwell using the single-cell manipulation platform equipped with an epi-fluorescence imaging mode. A detailed description is provided in the Supplementary materials and methods.
Fluorescence-assisted single cell harvesting
To distinguish and harvest different types of cells (CP-D/TurboGFP and EPC-2/FP635) from the co-culture in microwells, a fluorescence-assisted single cell harvesting platform was developed. To this end, a mercury arc epi-fluorescence illumination lamp and a cooled CCD camera were installed onto the pick-and-place single-cell manipulation platform equipped with appropriate excitation/emission filters. A Labview program was written for adjusting exposure time and gain settings of the fluorescent microscope. After cells were trypsinized as described in Single-cell collection section, they were visualized under transmitted light illumination. Co-cultured cells were then imaged with the camera in epi-fluorescence mode using the different filter cubes. CP-D/TurboGFP and EPC-2/FP635 cells can be easily distinguished in green and red channels, respectively. The two types of cells were then aligned and collected separately by the micropipette as described in the Single-cell collection section.
References
Lidstrom, M. E. & Meldrum, D. R. Life-on-a-chip. Nat Rev Microbiol 1, 158–164 (2003).
Eberwine, J. et al. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A 89, 3010–3014 (1992).
Elowitz, M. B., Levine, A. J., Siggia, E. D. & Swain, P. S. Stochastic gene expression in a single cell. Science 297, 1183–1186 (2002).
Irish, J. M. et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 118, 217–228 (2004).
Wang, D. & Bodovitz, S. Single cell analysis: the new frontier in ‘omics’. Trends Biotechnol 28, 281–290 (2010).
Arai, F. et al. On chip single-cell separation and immobilization using optical tweezers and thermosensitive hydrogel. Lab Chip 5, 1399–1403 (2005).
Kelbauskas, L. et al. Method for physiologic phenotype characterization at the single-cell level in non-interacting and interacting cells. J Biomed Opt 17, 0370081–03700812 (2012).
Anis, Y. H., Holl, M. R. & Meldrum, D. R. Automated selection and placement of single cells using vision-based feedback control. IEEE Trans Autom Sci Eng 7, 598–606 (2010).
Anis, Y., Houkal, J., Holl, M., Johnson, R. & Meldrum, D. Diaphragm pico-liter pump for single-cell manipulation. Biomed Microdevices 13, 651–659 (2011).
Tietjen, I. et al. Single-cell transcriptional analysis of neuronal progenitors. Neuron 38, 161–175 (2003).
Zare, R. N. & Kim, S. Microfluidic platforms for single-cell analysis. Annu Rev Biomed Eng 12, 187–201 (2010).
Mammoto, A., Mammoto, T. & Ingber, D. E. Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci 125, 3061–3073 (2012).
Roman, B. L. & Pekkan, K. Mechanotransduction in embryonic vascular development. Biomech Model Mechanobiol 11, 1149–1168 (2012).
Martins, R. P., Finan, J. D., Guilak, F. & Lee, D. A. Mechanical regulation of nuclear structure and function. Annu Rev Biomed Eng 14, 431 (2012).
Jean, C., Gravelle, P., Fournie, J. & Laurent, G. Influence of stress on extracellular matrix and integrin biology. Oncogene 30, 2697–2706 (2011).
Hynes, R. O. The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 (2009).
von Wichert, G. et al. RPTP-α acts as a transducer of mechanical force on αv/β3-integrin–cytoskeleton linkages. J Cell Biol 161, 143–153 (2003).
Jiang, G., Huang, A. H., Cai, Y., Tanase, M. & Sheetz, M. P. Rigidity Sensing at the Leading Edge through αvβ3 Integrins and RPTPα. Biophys 90, 1804–1809 (2006).
Yu, C.-h., Law, J. B. K., Suryana, M., Low, H. Y. & Sheetz, M. P. Early integrin binding to Arg-Gly-Asp peptide activates actin polymerization and contractile movement that stimulates outward translocation. Proc Natl Acad Sci U S A 108, 20585–20590 (2011).
Wang, H.-B., Dembo, M., Hanks, S. K. & Wang, Y.-l. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci U S A 98, 11295–11300 (2001).
Schober, M. et al. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol 176, 667–680 (2007).
Balda, M. S. & Matter, K. Epithelial cell adhesion and the regulation of gene expression. Trends Cell Biol 13, 310–318 (2003).
Dubash, A. D. et al. Focal adhesions: new angles on an old structure. Int Rev Cell Mol Biol 277, 1–65 (2009).
Roca-Cusachs, P., Iskratsch, T. & Sheetz, M. P. Finding the weakest link–exploring integrin-mediated mechanical molecular pathways. J Cell Sci 125, 3025–3038 (2012).
Phelan, M. C. Basic techniques in mammalian cell tissue culture. Curr Protoc Cell Biol 1.1. 1–1.1. 18 (2007).
Chaudhry, M. A. Induction of Gene Expression Alterations by Culture Medium from Trypsinized Cells. J Biol Sci 8, 81–87 (2008).
Zhang, X., Jones, P. & Haswell, S. J. Attachment and detachment of living cells on modified microchannel surfaces in a microfluidic-based lab-on-a-chip system. Chem Eng 135, S82–S88 (2008).
Chen, K. D. et al. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins and Shc. J Biol Chem 274, 18393–18400 (1999).
Flores, D., Battini, L., Gusella, G. L. & Rohatgi, R. Fluid shear stress induces renal epithelial gene expression through polycystin-2-dependent trafficking of extracellular regulated kinase. Nephron Physiol 117, p27–p36 (2010).
Zeng, J. et al. Quantitative single-cell gene expression measurements of multiple genes in response to hypoxia treatment. Anal Bioanal Chem 401, 3–13 (2011).
Park, N. J. et al. RNAprotect saliva: An optimal room-temperature stabilization reagent for the salivary transcriptome. Clin Chem 52, 2303–2304 (2006).
Fujita, J. Cold shock response in mammalian cells. J Mol Microbiol Biotechnol 1, 243–255 (1999).
Guzhova, I. & Margulis, B. Hsp70 chaperone as a survival factor in cell pathology. Int Rev Cytol 254, 101–149 (2006).
Murphy, M. E. The HSP70 family and cancer. Carcinogenesis 34, 1181–1188 (2013).
Wassarman, P. M. & Keller, G. M. Differentiation of embryonic stem cells. Vol. 365 (Elsevier, 2003).
Rasmussen, J. G. et al. Prolonged hypoxic culture and trypsinization increase the pro-angiogenic potential of human adipose tissue-derived stem cells. Cytotherapy 13, 318–328 (2011).
Zhu, H., Holl, M., Ray, T., Bhushan, S. & Meldrum, D. R. Characterization of deep wet etching of fused silica glass for single cell and optical sensor deposition. J Micromech Microeng 19, 065013 (2009).
Harada, H. et al. Telomerase Induces Immortalization of Human Esophageal Keratinocytes Without p16INK4a Inactivation. Mol Cancer Res 1, 729–738 (2003).
Palanca-Wessels, M. C. et al. Genetic analysis of long-term Barrett's esophagus epithelial cultures exhibiting cytogenetic and ploidy abnormalities. Gastroenterology 114, 295–304 (1998).
Lader, E. S. inventor; Applied Biosystems, Llc, assignee. Methods and reagents for preserving RNA in cell and tissue samples. United States patent US 8,178,296 B2. 2012 May 12.
Forman, S. & Jia, X. inventors; Zymo Research Corporation, assignee. Nucleic acid purification. United States patent US 20,120,184,725 A1. 2012 Jul 19.
Gao, W., Zhang, W. & Meldrum, D. R. RT-qPCR based quantitative analysis of gene expression in single bacterial cells. J Microbiol Methods 85, 221–227 (2011).
Acknowledgements
The authors thank Jordan Yaron for providing TurboGFP primers and Patti Senechal-Willis for providing cell-culture. This work was supported by the following grants to Dr. Deirdre R. Meldrum from the NIH National Human Genome Research Institute, Centers of Excellence in Genomic Sciences, Grant Numbers 5P50HG002360 and 3P50HG002360-10S1.
Author information
Authors and Affiliations
Contributions
J.Z. and A.M. performed experiments and data analysis, wrote the manuscript. W.G. performed experiments and data analysis. S.M. provided shear force calculations for the cell manipulation platform. D.S. developed and optimized instrument control software for cell harvesting. L.K. conceived and designed experiments, analyzed the data and wrote parts of the manuscript. D.M. provided overall study guidance and critically reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary materials
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zeng, J., Mohammadreza, A., Gao, W. et al. A Minimally Invasive Method for Retrieving Single Adherent Cells of Different Types from Cultures. Sci Rep 4, 5424 (2014). https://doi.org/10.1038/srep05424
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05424
This article is cited by
-
Isolation of primary brain endothelial cells, pericytes and astrocytes on a microfluidic immunopanning chip (MIC)
Microfluidics and Nanofluidics (2021)
-
Simultaneous Multiparameter Cellular Energy Metabolism Profiling of Small Populations of Cells
Scientific Reports (2018)
-
Platform for combined analysis of functional and biomolecular phenotypes of the same cell
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
