
Abstract
Introducing a point mutation is a fundamental method used to demonstrate the roles of particular nucleotides or amino acids in the genetic elements or proteins and is widely used in in vitro experiments based on cultured cells and exogenously provided DNA. However, the in vivo application of this approach by modifying genomic loci is uncommon, partly due to its technical and temporal demands. This leaves many in vitro findings un-validated under in vivo conditions. We herein applied the CRISPR/Cas9 system to generate mice with point mutations in their genomes, which led to single amino acid substitutions in proteins of interest. By microinjecting gRNA, hCas9 mRNA and single-stranded donor oligonucleotides (ssODN) into mouse zygotes, we introduced defined genomic modifications in their genome with a low cost and in a short time. Both single gRNA/WT hCas9 and double nicking set-ups were effective. We also found that the distance between the modification site and gRNA target site was a significant parameter affecting the efficiency of the substitution. We believe that this is a powerful technique that can be used to examine the relevance of in vitro findings, as well as the mutations found in patients with genetic disorders, in an in vivo system.
Similar content being viewed by others

Introduction
Introducing point mutations is a widely used experimental approach to evaluate the roles of specific nucleotides or amino acids in the function of the genetic elements or in proteins of interest. Using this technique, various nucleotides and amino acids have been proved to be indispensable for promoter or enhancer activity, as well as for the function or regulation of enzymes, transcription factors and signaling molecules. However, these findings were achieved mainly by in vitro experiments using exogenously provided DNA, such as plasmids or virus vectors and the in vivo introduction of defined point mutations at endogenous genomic loci has been uncommon, largely because of the technical and temporal costs associated with such techniques. Thus, the in vivo context or relevance of in vitro findings has remained unexplored in many cases.
The CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat/CRISPR-associated 9) system is a recently developed genome-engineering tool based on the bacterial CRISPR immune system, in which guide RNA (gRNA) recruits the Cas9 nuclease to the target locus in the genome by sequence complementarity and induces double strand breaks (DSBs)1,2. These DSBs cause small insertions or deletions (indels) following non-homologous end-joining (NHEJ) repair, or can be utilized to introduce defined sequence modifications through a homology-dependent repair (HDR) mechanism1,2. While the use of HDR-dependent genomic engineering has been reported in in vitro cell culture systems, whether this could be applied in in vivo systems to introduce defined point mutations and what parameters affect its efficiency were less explored.
In the present study, we applied the CRISPR/Cas9 system to create a single amino acid substituted mouse model. By microinjecting synthesized RNAs and single-strand oligodeoxynucleotide (ssODN) donor sequences into mouse zygotes, we were able to introduce defined point mutations in the mouse genome, which led to single amino acid substitutions in the proteins of interest, within as short as one to two months. Using this technique, we can now evaluate the in vivo relevance of particular nucleotides/amino acids with a low cost and within a short time, which facilitates the elucidation of the in vivo context of the findings in cultured cell systems, as well as the in vivo screening for the relevant mutation among those found in human patients with various diseases.
Results
Substitution by single gRNA and wild-type hCas9
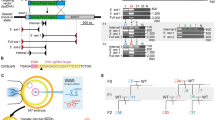
To generate an amino acid substituted mouse model using the CRISPR system, we first took the simplest approach: microinjecting single gRNA, wild type (WT) hCas9 mRNA and a ssODN with a single base mismatch to the genomic sequence, which caused single amino acid conversion, into mouse zygotes. As a target, a c.274C>T mutation of the Steroidogenic Factor 1 gene (Sf-1, also known as Ad4BP or Nr5a1)3, which causes a p.R92W amino acid substitution in the SF-1 protein, was chosen and gRNAs and ssODNs were designed as shown in Figure 1A.

Introduction of the C>T substitution in Sf-1 locus.
(A) A schematic illustration showing the locations of the gRNAs and ssODNs, along with the mouse Sf-1 locus. Blue bars and letters indicate the position of the gRNA targets with red bars highlight the PAM sequences. ssODNs are shown in green letters at the corresponding position to the Sf-1 locus. Red letters indicate the substitution target site in the Sf-1 locus and corresponding mismatched nucleotides in the ssODNs. The deduced amino acid sequences from wild type (WT) and substituted sequences are shown at the bottom, with red letters indicating the target and the results of the substitution. (B) The results of the sequence analysis of the Sf-1 locus of a representative mouse, with a monoallelic C>T substitution. The Sf-1 locus around the gRNA target site was PCR amplified and directly sequenced (Upper panel), or cloned into the plasmid and sequenced independently (Lower panel). The upper panel shows the electropherogram of the direct sequencing results, in which one allele received a C>T substitution. WT and donor DNA sequences are shown on the top, with red letters indicating the mutated nucleotide. The red arrow indicates the overlapping peak caused by the monoallelic substitution. The lower panel shows the sequence alignment of eight independent clones. The eight sequences are separated into two types: the WT sequence and substituted sequences. (C) The results of the direct sequencing of the Sf-1 locus around the gRNA target site of another representative mouse obtained in experiment 4 shown in Table 1, in which one alleles received C>T substitutions, followed by additional mutation(s) that caused continuous overlapping peaks.
To examine whether the sequence or location of the gRNAs could affect the efficiency of the substitution, we designed three independent gRNAs (Sf-1gRNA1, 2 and 3), which had their Protospacer-Adjacent Motif (PAM) sequence 52 or 7 bases upstream, or 12 bases downstream, of the target point mutation (Fig. 1A). Three independent ssODNs were also designed, placing the mismatched nucleotide or PAM sequence of gRNA1 and gRNA2 in their center, respectively (Fig. 1A).
Various combinations of gRNAs and ssODNs (Table 1) were mixed with hCas9 mRNA and microinjected into pronuclear stage embryos. The injected zygotes were transferred to pseudo-pregnant females at the two-cell stage and the obtained pups were examined for the sequence around the Sf-1 locus. As shown in Table 1, we found total 26 mice with the C>T substitution generated from three out of four experiments (experiments 2, 3 and 4) and 7 of them had biallelic substitutions (Fig. 1B and Table 1). These results show that microinjecting gRNA, WT hCas9 mRNA and the ssODN donor into mouse zygotes is sufficient to introduce a defined single base substitution in their genome.
Of note, while the frequencies of DSB caused by hCas9/gRNAs, which are indicated by the frequencies of mutated alleles, are comparable among 4 experiments (68–89%, P = 0.75) (Table 1), the frequencies of designated substitutions vary significantly (0–29.6%, P < 0.01) (Table 1). This lack of a correlation between the frequency of the mutation and the substitution to occur suggests that there may be an optimal rate of DSB occurance to introduce HDR-dependent sequence incorporation (such like too frequent DSB might be unfavorable for HDR), or that the distance and/or the mutual position between the substitution target site and gRNA PAM sequences (surrogate DSB point) could affect the substitution efficiency. Our subsequent experiments support the latter possibility (see below). To explore the effect of mutual position between substitution target site and DSB point on substitution efficiency, we designed two additional ssODNs which have similar sequence to ssODN1, but carry point mutations in 36 base upstream or 12 base downstream to the PAM sequence of Sf-1 gRNA3 (Supplementary Fig. S1). ssODNs were microinjected into mouse zygotes with hCas9 mRNA and Sf-1 gRNA3 and the frequency of the designated substitution and the mutated allele in those mice were compared to experiment 4 (Supplementary Table S1). As a result, while 80–95% of mice had mutated alleles in all three experiments, the frequency of the substituted alleles differs substantially: 29.6% with ssODN3, 20.0% with ssODN4 and 0% with ssODN5 (Supplementary Table S1). These results indicate that the probability of the mutation in donor DNA to be incorporated into the genome declines with the distance between the DSB point and the mutation. In addition, the mutation which is at the downstream (3′ side) of PAM sequence showed slightly lower frequency of substitution than the mutation with the same distance from the PAM sequence but is at the upstream (5′ side). Together, these results show that the mutual location of gRNA target site and substitution target site is an important parameter for the efficient introduction of the designated point mutation.
Although there was an efficient generation of targeted point mutations, we observed that many mice with the C>T substitution also received additional random mutations in the flanking regions (Fig. 1C and Table 1). Moreover, it has previously been reported that single gRNA/WT hCas9 may introduce undesired mutations at the genomic loci with similar sequences to the target site (off-target effect)4,5,6, which prompted us to test the double nicking strategy5 to induce more accurate genome modifications in mouse zygotes.
Substitution using two gRNAs and hCas9 D10A (double nicking)
Combining a pair of gRNAs with mutated hCas9 (hCas9 D10A), double nicking cause DSB only at the genomic loci where both of the gRNAs bind at an appropriate distance, thus making it highly specific to the target site5. To examine whether this set-up could be used for the HDR-dependent genome editing in vivo, we aimed to induce the c.1187A>G mutation of the Sox9 gene, which results in a p.K396R amino acid substitution in the SOX9 protein. Lysine 396 is an evolutionary-conserved post-translational modification site of the SOX9 protein, but its in vivo relevance has not been fully examined7,8. We designed two gRNAs with a five-base offset where their PAM sequences are positioned about 40 bases upstream and three bases downstream of the A>G substitution site (Fig. 2A). Two ssODNs were designed with several silent mutations within the recognition sequence of gRNA2 to avoid repetitive digestions. An additional silent mutation to eliminate the PvuII site was also designed to facilitate the genotyping (Fig. 2A). Sox9-ssODN1 causes both silent mutations and c.1187A>G substitution (K-to-R substitution), while Sox9-ssODN2 causes only silent mutations (K-to-K substitution) (Fig. 2A, lower panel). The two gRNAs, hCas9 D10A mRNA and 2 ssODNs were mixed and microinjected into zygotes to generate amino acid substituted mice (by Sox9-ssODN1) and negative control mice (by Sox9-ssODN2) simultaneously.

Introduction of the K396R substitution in Sox9 locus.
(A) A schematic illustration demonstrating the locations of the gRNAs and ssODNs along with the mouse Sox9 locus. The blue bars and letters indicate the positions of gRNA targets, while red bars highlight the PAM sequences. The sequences of ssODNs are shown in green letters at the corresponding positions to the Sox9 locus. Red letters indicate the substitution target site in the Sox9 locus and the corresponding mismatched nucleotides in the ssODNs. The deduced amino acid sequences from wild type (WT) and substituted sequences are shown at the bottom, with red letters indicating the target and the results of the substitution. Orange letters in the sequences are the targets of silent mutations, which do not cause amino acid conversion. The PvuII site is underlined in the Sox9 loci which had mutated in ssODNs. (B) The results of the sequence analysis of the Sox9 locus of a representative mouse, which had a monoallelic K-to-R substitution. The Sox9 locus around the gRNA target site was PCR amplified and directly sequenced (Upper panel), or was cloned into the plasmid and sequenced independently (Lower panel). The upper panel shows the electropherogram of the direct sequencing result of a mouse obtained in experiment 1 shown in Table 2, in which one allele received a K-to-R substitution. The WT and donor DNA sequences are shown on the top, with orange and red letters indicating mutated nucleotides. The orange and red arrows indicate the overlapping peaks caused by monoallelic substitutions and a black arrow indicates the non-overlapping peak despite the fact that the ssODN (donor DNA) had a mutated nucleotide. The lower panel shows the sequence alignment of seven independent clones. The seven sequences were separated into two types: the WT and substituted sequences.
The obtained pups were genotyped as before and we found that five pups out of 24 (20.8%) with the designated substitution, two of which had a K-to-R substitution (Fig. 2B) and three of which had a K-to-K substitution (Table 2). A frame shift mutation caused by an indel was observed in only one mouse. Thus, double nicking, together with donor ssODN, can also introduce defined point mutations in mouse zygotes. The frequency of the substitution was lower than that of the single gRNA/WT hCas9 condition shown in previous section, but was sufficient to generate mice with two genotypes in a single microinjection experiment by mixing different donor DNAs. Of note, a PvuII site substitution was never observed in the mice with upstream substitutions (Fig. 2B and data not shown). This, together with the results of the experiment 4–6 with Sf-1, suggest that only a part of the sequence of the donor DNA near the DSB site will be incorporated through the CRISPR-mediated HDR pathway. Therefore, it is important to place the gRNA target site close enough to the substitution site to achieve efficient substitution.
SOX9-K396R gRNA2 and WT hCas9 mRNA with ssODNs were also microinjected into zygotes to compare the efficiency with double nicking (Table 2). As shown in Table 2, single gRNA with WT hCas9 resulted in a higher mutation frequency (76.9%) as well as substitution frequency (34.6%) and as in the case of Sf-1, biallelic substitution and additional random mutations were also observed (Supplementary Fig. S2 and Table 2).
Finally, we compared the off-target effects under the two conditions. Five putative off-target sites of SOX9-K396R gRNA2 were computationally predicted6 and the sequences of those sites in gRNA/hCas9-injected mice were analyzed (Fig. 3A). As a result, we did not detect any mutations in the total of 250 loci (five off-target sites in 50 mice) examined (Fig. 3B and Table 3), indicating that both single gRNA with WT hCas9 and double nicking introduced designated genetic modification in highly specific manner. To examine if this high specificity could also be observed with other gRNAs, off-target effect of Sf-1 gRNAs were also analyzed. We focused on the mice which received designated substitution in first 4 experiments (total 26 mice shown in Table 1) and examined total of 121 loci and did not find any off-target mutation (Supplementary Fig. S3 and Supplementary Table S2). Together, these results suggest the ratio of on/off-target mutation is substantially high in this experimental set up.

Off-target analysis of SOX9-K396R gRNAs.
(A) The sequences and positions of possible off-target sites for SOX9-K396R gRNA2. OT: Off-target site. (B) The results of the sequence analysis of the target site and off-target sites. Direct sequencing of the PCR-amplified Sox9 locus and five putative off-target sites from single representative mouse are shown as representative results. Only the sequence of the Sox9 locus showed overlapping peaks caused by substitution. The gRNA target and possible off-target sequences were shown at the fourth to twenty-sixth bases, as indicated by the darker background color.
Discussion
Introducing point mutations is widely used technique in cultured cell based experiments, but uncommon in in vivo system partly because of its temporal and technical demands. The standard approach to examine the function of particular nucleotides or amino acids in in vivo background has been to create a knock-in or transgenic mouse that carries cDNA or other genetic element with defined point mutation(s). However these techniques require long and laborious procedures such as constructing targeting vectors, cloning of ES cell and/or establishing multiple transgenic mouse lines. Our method overcome those difficulties and possible problems and is ideal to observe the effect of subtle genetic modification under physiological condition.
The CRISPR/Cas9 system has been used to introduce defined genetic modification in cultured cell system5,9, but whether this could be applied to in vivo system and what parameter(s) is influential for its efficiency was less explored. We demonstrated that the CRISPR/Cas9 system, both single gRNA/WT hCas9 and double nicking set-ups, was effective in mouse zygotes with the former having slightly higher efficiency. In our best condition, microinjection of single gRNA with WT hCas9 introduced the point mutation in more than 30% of mice examined, in which 10% of them received biallelic modification, allowing researcher to analyze the mouse model in the founder generation (Note that the possible mosaicism should be considered in analyzing the phenotype of founder generation and we believe that the confirmation in descendant generation is indispensable to conclude genotype-phenotype relationship). We observed higher efficiency with single gRNA with WT hCas9 than double nicking set-up, which is expected because the latter set-up requires two gRNA-hCas9 complexes to be recruited simultaneously to the target site to introduce DSB while one complex is enough for the former. In addition, to inject the same amount of RNAs in total, the concentration of each gRNA and hCas9 mRNA was lower in our double nicking set-up, which might also affect the efficiency (see Methods for the details).
The mutual position of substitution target sites and gRNA target sites (DSB points) seems to affect the substitution efficiency. Using same gRNA and ssODNs with various point mutations, we found that the mutations which are closer to the gRNA target site is incorporated more often than that are farther from DSB point (Supplementary Fig. S1 and Supplementary Table S1). This is in line with the previous report in which the efficiency of gene conversion was examined using plasmid donor DNA and restriction enzyme in cell culture system and found that the frequency of the substitution declines with the distance from the digestion site10. Whether the frequency of substitution varies by using sense (i.e. the same strand of gRNA target) or anti-sense strand sequence for ssODN could be another point to be examined in the future studies.
Surprisingly, we did not observe any off-target effect in our experiments. The total of 371 loci were examined but no indels were detected. This was an unexpected result, since frequent off-target mutagenesis has been reported in human cells4,5, but is consistent with a rare off-target effect observed in mouse zygotes microinjected with hCas9 and gRNA plasmids11. These results do not exclude off-target effects in general, but imply that the cultured cells and mouse zygotes have distinct risks for off-target effects, possibly due to the different stability of the CRISPR/Cas9 components and/or the nuclear/chromatin dynamics between stable cell lines and zygotes under early developmental process. Further accumulation of data is necessary to understand the kinetics of CRISPR mediated genome modification to optimize the experimental parameters for obtaining highest efficiency with minimum off-target effect in various back ground.
In summary, we have established an experimental system to introduce defined point mutations into the mouse genome using the CRISPR/Cas9 system, which enabled us to generate an amino acid substituted mouse model at a low cost and in a short time. The WT hCas9 and double nicking strategy were both effective and the distance between the DSB site and substitution site seems to be an important parameter for determining the substitution efficiency. We believe this technique will be useful to reveal the in vivo relevance of particular nucleotides/amino acids identified in in vitro systems, as well as to reproduce the genetic variations found in patients of genetic disorders in mouse models.
Methods
Plasmids
hCas9, hCas9 D10A and the gRNA cloning vector1 were purchased from Addgene (Plasmid ID #41815, #41816 and #41824, respectively). Since the original gRNA cloning vector lacks the partial sequence of the U6 promoter and gRNA scaffold, we first modified it by adding the following sequence: 5′-GTGGAAAGGACGAAACACCGGCTAGCAGGCCTATCGATGTTTTAGAGCTAGAAATAGC-3′ into the AflII site to fill the missing sequence and to facilitate further cloning. gRNA expression vectors were constructed by inverse PCR using this modified vector and the primer pairs shown in the Table below. The PCR products were DpnI digested and used for the transformation of E. coli (DH5a). The sequences of the obtained constructs were validated by sequencing.
gRNA design
The gRNAs were designed by searching for “GG” or “CC” sequences near the point mutation target sites and were defined as N(21)GG or the reverse complement sequence of CCN(21). The gRNA pairs used for double-nicking were designed by searching for CCN(40–48)GG sequences and the pair closest to the substitution site was chosen.
The gRNAs used in this study were: Sf-1 gRNA1 5′-GTGGGCAGGAGCAGTCTGTCAGG-3′, Sf-1 gRNA2 5′-GTGCGTGCTGATCGAATGCGGGG-3′, Sf-1 gRNA3 5′-GGGGTGGCCGGAACAAGTTTGGG-3′, SOX9-K396R gRNA1 5′-GCCTGGCTCGCTGCTCAGCGTGG-3′, SOX9-K396R gRNA2 5′-CCAGCGAACGCACATCAAGACGG-3′.
RNA synthesis
Template DNAs for in vitro transcription were generated by PCR amplification of the ORFs of hCas9 and hCas9 D10A, as well as gRNAs (i.e. guide sequences and scaffold sequences) from each plasmid using PrimeSTAR (TaKaRa) and the primer sequences shown below (the T7 RNA polymerase recognition sequence was attached to the 5′ end of the fw primers). The PCR products were purified and used for the in vitro RNA synthesis using mMessage mMachine T7 kit (lifetechnologies). The reaction scale was doubled and the reaction time was extended to over-night to obtain a sufficient amount of gRNA. The synthesized RNAs were purified using an RNeasy mini kit (Qiagen) with an additional ethanol precipitation.
ssODN
The synthesized single stranded oligonucleotides (110 bases, PAGE purified) were purchased from fasmac.
Sf-1 ssODN1 5′-GACTGCTCCTGCCCACAGCTGTGCGTGCTGATCGAATGCGGGGTGGCTGGAACAAGTTTGGGCCCATGTACAAGAGAGACCGGGCCTTGAAGCAGCAGAA-3′, Sf-1 ssODN2 5′-TTTGGGAAAAGATCTGTGGCAGACAGTTGGGAAGGTAGAACCTGACAGACTGCTCCTGCCCACAGCTGTGCGTGCTGATCGAATGCGGGGTGGCTGGAAC-3′, Sf-1 ssODN3 5′-TGGGAAGGTAGAACCTGACAGACTGCTCCTGCCCACAGCTGTGCGTGCTGATCGAATGCGGGGTGGCTGGAACAAGTTTGGGCCCATGTACAAGAGAGAC-3′, Sf-1 ssODN4 5′-GACTGCTCCTGCCCACAGCTGTGCGTGCTGATCGAATGCGGGGTGGCCGGAACAAGTTTGGGCCCATGTATAAGAGAGACCGGGCCTTGAAGCAGCAGAA-3′, Sf-1 ssODN5 5′-GACTGCTCCTGCCCACAGCTGTGTGTGCTGATCGAATGCGGGGTGGCCGGAACAAGTTTGGGCCCATGTACAAGAGAGACCGGGCCTTGAAGCAGCAGAA-3′, Sox9-K396R ssODN1 5′-ACACACGCTCACCACGCTGAGCAGCGAGCCAGGCCAGTCCCACAGGACCCATATCAGGACGGAGCAACTGAGCCCCAGCCACTACAGCGAGCAGCAGCAGCACTCCCCGC-3′, Sox9-K396R ssODN2 5′-ACACACGCTCACCACGCTGAGCAGCGAGCCAGGCCAGTCCCACAGGACCCATATCAAGACGGAGCAACTGAGCCCCAGCCACTACAGCGAGCAGCAGCAGCACTCCCCGC-3′.
Microinjection
The microinjection of mouse zygotes was performed as described before12,13. Essentially, mouse zygotes were obtained by mating superovulated BDF1 females and WT BDF1 males (Sankyo lab service). RNAs and ssODNs were mixed just before microinjection into the cytoplasm or pro-nuclei of zygotes and the injected embryos were incubated at 37°C until they were transferred into pseudo-pregnant females at the two-cell stage. The concentration of injected RNAs was always kept at 500 ng/μl in total. For the single gRNA/WT Cas9 condition, gRNA and hCas9 mRNA were mixed at a 1:1 ratio and thus the final concentration was 250 ng/μl each and for the double nicking condition, the gRNAs and hCas9 mRNA were mixed at 1:1:1 ratio and thus a final concentration of 167 ng/μl each. The concentration of injected ssODNs was final 100 ng/μl. The protocols for animal experiments were approved by the Animal Care and Use Committee of the National Research Institute for Child Health and Development (Permit Numbers: A2004-003-C09, A2009-002-C04).
Genotyping
Genomic DNA was extracted from the tail tips of pups and the genomic sequences around the gRNA target sites were PCR amplified using the primers shown below. The obtained PCR products were treated by ExoSAP-IT (USB) and sequenced directly, or were cloned into the plasmid and sequenced.
The criteria for the “mutation” and “substitution”
The genomic sequences around the gRNA target sites were PCR amplified and served for direct sequencing (see above). The electropherograms of each sequence were classified into three categories: wild type (no overlapping peaks), mutated (continuous overlapped peaks, which started from the gRNA target site) and substituted (overlapped peak(s) at the designated position(s)) (Supplementary Fig. S2). The sequences were also aligned with the wild type sequence to check whether there was a homozygous mouse with identical indels on both alleles (which also produce a no overlapping peak pattern). PCR products were cloned into the plasmid and multiple clones were sequenced to determine the sequences of each allele when necessary.
Off-target analysis
Possible off-target sites for SOX9-K396R gRNA2 and Sf-1 gRNA 1, 2 and 3 were predicted by an online-based tool (http://www.genome-engineering.org/)6. Four or five putative off-target sites were found allowing a maximum of three to four bases of mismatches (Fig. 3A and Supplementary Fig. S3). The off-target sites were PCR amplified from the genomic DNA of gRNA-injected mice and analyzed by direct sequencing.
Primers
The primer sequences used in this study are shown below:
For vector construction: gRNA vector adopter fw 5′-GCTAGCAGGCCTATCGATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATC-3′, gRNA vector adopter rev 5′-ATCGATAGGCCTGCTAGCCGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAATCGAAATAC-3′.
For gRNA cloning: SOX9-K396R gRNA1 fw 5′-CCTGGCTCGCTGCTCAGCGGTTTTAGAGCTAGAAATAGCAAG-3′, SOX9-K396R gRNA1 rev 5′-AACCGCTGAGCAGCGAGCCAGGCGGTGTTTCGTCCTTTCCAC-3′, SOX9-K396R gRNA2 fw 5′-CAGCGAACGCACATCAAGAGTTTTAGAGCTAGAAATAGCAAG-3′, SOX9-K396R gRNA2 rev 5′-AACTCTTGATGTGCGTTCGCTGCGGTGTTTCGTCCTTTCCAC-3′, Sf-1-gRNA1 fw 5′-TGGGCAGGAGCAGTCTGTCGTTTTAGAGCTAGAAATAGCAAG-3′, Sf-1-gRNA1 rev 5′-AACGACAGACTGCTCCTGCCCACGGTGTTTCGTCCTTTCCAC-3′, Sf-1-gRNA2 fw 5′-TGCGTGCTGATCGAATGCGGTTTTAGAGCTAGAAATAGCAAG-3′, Sf-1-gRNA2 rev 5′-AACCGCATTCGATCAGCACGCACGGTGTTTCGTCCTTTCCAC-3′, Sf-1-gRNA3 fw 5′-GGGTGGCCGGAACAAGTTTGTTTTAGAGCTAGAAATAGCAAG-3′, Sf-1-gRNA3 rev 5′-AACAAACTTGTTCCGGCCACCCCGGTGTTTCGTCCTTTCCAC-3′.
For RNA synthesis:
T7-Sf-1-gRNA1 5′-TTAATACGACTCACTATAGGTGGGCAGGAGCAGTCTGTC-3′, T7-Sf-1-gRNA2 5′-TTAATACGACTCACTATAGGTGCGTGCTGATCGAATGCG-3′, T7-Sf-1-gRNA3 5′-TTAATACGACTCACTATAGGGGGTGGCCGGAACAAGTTT-3′, T7-SOX9-K396R gRNA1 5′-TTAATACGACTCACTATAGGCCTGGCTCGCTGCTCAGCG-3′, T7-SOX9-K396R gRNA2 5′-TTAATACGACTCACTATAGGCAGCGAACGCACATCAAGA-3′, gRNA rev template for RNA synthesis 5′-AAAAGCACCGACTCGGTGCC-3′, T7-hCAS9 5′-TAATACGACTCACTATAGGGAGAATGGACAAGAAGTACTCCATTGG-3′, hCAS9-rev 5′-TCACACCTTCCTCTTCTTC-3′.
For genotyping: Sox9 exon 3 fw 5′-ACCAATACTTGCCACCCAAC-3′, Sox9 exon 3 rev 5′-CGGCTGCGTGACTGTAGTAG-3′, Sf-1 exon 4 fw 5′-TGGGGAATGGTATAAGCGTGTG-3′, Sf-1 exon 4 rev 5′-CGTGCAGGCTAGGGGGTAAC-3′.
For the off-target analysis:
SOX9-K396R gRNA2 OT1 fw 5′-TACACACCCGAGTCCCTTTC-3′, SOX9-K396R gRNA2 OT1 rev 5′-ACCAAACAACACGGCCTTAG-3′, SOX9-K396R gRNA2 OT2 fw 5′-ATCTGACTTGGCGTGGAAAC-3′, SOX9-K396R gRNA2 OT2 rev 5′-GACCAGGACCTGTCGTCAAT-3′, SOX9-K396R gRNA2 OT3 fw 5′-GCCAAGAGGAGGTAGCAGTG-3′, SOX9-K396R gRNA2 OT3 rev 5′-CACATCCCCATAGGAAATGG-3′, SOX9-K396R gRNA2 OT4 fw 5′-GTCTCTTGGCCTCTGCAATC-3′, SOX9-K396R gRNA2 OT4 rev 5′-AGCTCCACCCACAGAGAGAA-3′, SOX9-K396R gRNA2 OT5 fw 5′-AGACAGAGCTGCTGCAAACA-3′, SOX9-K396R gRNA2 OT5 rev 5′-TGTCAACTACCGCACATGGT-3′, Sf-1 gRNA1 OT1 fw 5′-AGCAGAGAAGCAGGAGCAAG-3′, Sf-1 gRNA1 OT1 rev 5′-CCATCCCAAACTCAGCTGTT-3′, Sf-1 gRNA1 OT2 fw 5′-CTTGACTGCTTCTGGGAAGG-3′, Sf-1 gRNA1 OT2 rev 5′-GCCCACCTGTGCCTTATCTA-3′, Sf-1 gRNA1 OT3 fw 5′-GACCATTGGAGGGCAAACTA-3′, Sf-1 gRNA1 OT3 rev 5′-GGCCAGGCTATAAACACCAA-3′, Sf-1 gRNA1 OT4 fw 5′-AGTGTGAGGTCCCTGTTTGG-3′, Sf-1 gRNA1 OT4 rev 5′-AGGCTTAGGGATCTGGCATT-3′, Sf-1 gRNA1 OT5 fw 5′-CTTGCCTTTTCTCTGCCATC-3′, Sf-1 gRNA1 OT5 rev 5′-GCAGCCTGAAGGAAATGAAG-3′, Sf-1 gRNA2 OT1 fw 5′-TGGGAGAGTTCCCTGATTTG-3′, Sf-1 gRNA2 OT1 rev 5′-GTCTCCAAACCCACCAGAAA-3′, Sf-1 gRNA2 OT2 fw 5′-ACTCCCTGGGACACTGTCTG-3′, Sf-1 gRNA2 OT2 rev 5′-TGCAAGAGTCCACACTACGG-3′, Sf-1 gRNA2 OT3 fw 5′-TTCCTAAGTTGGCTCGCAGT-3′, Sf-1 gRNA2 OT3 rev 5′-CTTGGGTTTTTCATGGGCTA-3′, Sf-1 gRNA2 OT4 fw 5′-GATTAAAGGCGTGTGCCACT-3′, Sf-1 gRNA2 OT4 rev 5′-TCCCAGCCACATTCATGTTA-3′, Sf-1 gRNA3 OT1 fw 5′-GCACCGTCAAGGAGAAAGAG-3′, Sf-1 gRNA3 OT1 rev 5′-GTTCCCCAGTCCTTCAACAA-3′, Sf-1 gRNA3 OT2 fw 5′-CACCAATTCAGCGACATCAG-3′, Sf-1 gRNA3 OT2 rev 5′-AAAGATGCTTGCACCTTGCT-3′, Sf-1 gRNA3 OT3 fw 5′-TGAGTCCAGCCTCATCACAG-3′, Sf-1 gRNA3 OT3 rev 5′-CCCTGTCTCCAAACACCCTA-3′, Sf-1 gRNA3 OT4 fw 5′-GCTTGTCATGCTCAGTTCCA-3′, Sf-1 gRNA3 OT4 rev 5′-GACCTTTGGAAGAGCAGTCG-3′, Sf-1 gRNA3 OT5 fw 5′-TCCAGGAGACCTGTTGCTCT-3′, Sf-1 gRNA3 OT5 rev 5′-AGGCTCCAAGACAGTGTGGT-3′.
Statistical analysis
Statistical analysis to assess the differences of the frequency of mutations and substitutions among experiments 1–4 (Table 1) was performed using the χ2 test. We hypothesized that the expected frequency (calculated as total number of mice with mutated (or substituted) allele/total number of genotyped mice) is same among 4 experiments and esamined with χ2 test.
References
Mali, P. et al. RNA-Guided Human Genome Engineering via Cas9. Science 339, 823–826 (2013).
Cong, L. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823 (2013).
Parker, K. L. & Schimmer, B. P. Steroidogenic factor 1: a key determinant of endocrine development and function. Endocrine Reviews 18, 361–377 (1997).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–6 (2013).
Ran, F. A. et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 154, 1380–9 (2013).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Akiyama, H. et al. The transcription factor Sox9 is degraded by the ubiquitin-proteasome system and stabilized by a mutation in a ubiquitin-target site. Matrix Biol. 23, 5083–5093 (2005).
Lee, P. C. et al. SUMOylated SoxE factors recruit Grg4 and function as transcriptional repressors in the neural crest. J. Cell Biol. 198, 799–813 (2012).
Wang, H. et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 153, 910–918 (2013).
Elliott, B., Richardson, C., Winderbaum, J., Nickoloff, J. A. & Jasin, M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cell Biol. 18, 93–101 (1998).
Mashiko, D. et al. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 3, 3355 (2013).
Kato, T. et al. Production of Sry knockout mouse using TALEN via oocyte injection. Sci. Rep. 3, 3136 (2013).
Takada, S. et al. Targeted Gene Deletion of miRNAs in Mice by TALEN System. PLoS ONE 8, e76004 (2013).
Acknowledgements
This work was supported in part by the Grant from the National Center for Child Health and Development (Grant Number 25-1 for M. Inui and 24-3 for S.T.), Japan Society for the Promotion of Science KAKENHI (Grant Number 25871177 for M. Inui and Grant Number 25132713 for S.T.), the Ministry of Education, Culture, Sports, Science and Technology KAKENHI (Grant Number 23570265) to S.T.
Author information
Authors and Affiliations
Contributions
M. Inui designed the substitution of Sox9 and M. Igarashi. and M.F. defined the position of Sf-1 substitution. A.K. constructed gRNA vectors. M.T. performed the microinjection. M. Inui, M.M. and S.Y. carried out the genotyping and the sequence analysis. M. Inui, H.A. and S.T. designed the project and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Inui et al. Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Inui, M., Miyado, M., Igarashi, M. et al. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci Rep 4, 5396 (2014). https://doi.org/10.1038/srep05396
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05396
This article is cited by
-
A novel mucopolysaccharidosis type II mouse model with an iduronate-2-sulfatase-P88L mutation
Scientific Reports (2023)
-
Strategies for generation of mice via CRISPR/HDR-mediated knock-in
Molecular Biology Reports (2023)
-
Bex1 is essential for ciliogenesis and harbours biomolecular condensate-forming capacity
BMC Biology (2022)
-
Modification of improved-genome editing via oviductal nucleic acids delivery (i-GONAD)-mediated knock-in in rats
BMC Biotechnology (2021)
-
Generation of scalable cancer models by combining AAV-intron-trap, CRISPR/Cas9, and inducible Cre-recombinase
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
