
Abstract
Spectrins and plakins are important communicators linking cytoskeletal components to each other and to cellular junctions. Microtubule-actin cross-linking factor 1 (MACF1) belongs to the spectraplakin family and is involved in control of microtubule dynamics. Complete knock out of MACF1 in mice is associated with developmental retardation and embryonic lethality. Here we present a family with a novel neuromuscular condition. Genetic analyses show a heterozygous duplication resulting in reduced MACF1 gene product. The functional consequence is affected motility observed as periodic hypotonia, lax muscles and diminished motor skills, with heterogeneous presentation among the affected family members. To corroborate these findings we used RNA interference to knock down the VAB-10 locus containing the MACF1 homologue in C. elegans and we could show that this also causes movement disturbances. These findings suggest that changes in the MACF1 gene is implicated in this neuromuscular condition, which is an important observation since MACF1 has not previously been associated with any human disease and thus presents a key to understanding the essential nature of this gene.
Similar content being viewed by others


Introduction
The cytoskeleton is a key factor in basic functions common to all cell types. It defines the shape and size of a given cell through its highly organized structure. In eukaryotic cells the cytoskeleton consists of 3 filament types: F-actin microfilaments, intermediate filaments such as vimentin, keratin and desmin and polymeric alpha- and beta-tubulin composing the microtubules. The cytoskeleton is dynamic and responds to environmental signalling cues1. Transfer of information into the cells occurs via cytoskeletal proteins such as spectrins and plakins, linking cytoskeletal components to each other and to junctions2. Microtubule-actin cross-linking factor 1 (MACF1, Online Mendelian Inheritance in Man (OMIM) database number 608271) is a member of the spectraplakin family3 and is a ubiquitously expressed 614 kDa protein4. MACF1 is distributed in a cytoplasmic filamentous network where it controls microtubule dynamics. MACF1 strengthens the link between F-actin and microtubules thereby facilitating polarization of cells and coordinated cellular movements5,6,7. The phenotype of MACF1-/- mice is developmental retardation and embryonic death at gastrulation stage e7.58, with a “head without trunk” phenotype, which has similarities to Wnt3-/- mice9. In zebra fish, a deletion mutation in the MACF1 homologous gene magellan caused failure in oocyte polarization10. These observations suggest a fundamental role for MACF1 in embryonic development and until now, there have been no reports of any disease linked to mutations in MACF1 likely due to the essential nature of this gene2,11.
Noticeably, MACF1 displays structural similarity to dystonin and dystrophin and has been considered a hybrid between the two4. However, another study has shown that one of the MACF1 isoforms contains 8 plectin repeats, thus this protein also displays structural similarity to plectin and has further been described as a hybrid between plectin and dystrophin12. Therefore this protein has features of several cytoskeletal proteins. Mutations in dystrophin cause the severe to mild Duchenne and Becker muscular dystrophies13, mutations in dystonin cause dystonia musculorum14 and mutations in plectin cause myopathy11 suggesting that MACF1 could be related to undiagnosed myopathic phenotypes.
Here we describe a novel myopathy with muscle weakness associated with reduced expression of MACF1 caused by a duplication of part of the MACF1 gene locus. We have named the condition spectraplakinopathy type 1 based on MACF1 belonging to the spectraplakin protein family and since there are more proteins in this family we have sub-classed the myopathy as type 1.
Results
Clinical presentation
The proband (subject III:2, figure 1a) is a 12-year old boy and the second child of a healthy father and a mother with mild hypotonic symptoms. He has been attended to by the health care system since birth. At the age of two and a half he was seen by a physiotherapist who described him as hypotonic and hypermobile with oligo-mimic and decreased endurance. He continuously has trouble chewing and eating as well as walking. His muscles are now weak with lax muscle tone and he is quickly exhausted when performing physical activity. At the age of 9 he had light contractures in his foot-, knee- and hip- joints. His skeletal muscle problems mainly manifest as generalized hypotonia and pain and the pain is induced by overload where the muscles become hard and sore. He has normal cardiac function (normal ECG). Routine laboratory tests and metabolic workup revealed no abnormalities and showed a normal Creatine Kinase (85–125 U/l, normal range 50–400 U/l) and Lactate Dehydrogenase (214 U/l, normal range 100–345 U/l) value. Neurological examinations showed no ptosis, normal pupil reactions and normal eye movements and a neurophysiological examination using electromyography showed normal motoric potentials with no indications of denervation. He has normal reflexes and normal muscle mass but a weak balance with reduced muscle tone. Extensive clinical examinations and screens for e.g. spinal muscular atrophy type II and III, cystic fibrosis, dystonia musculorum, Fabry's and Pompe's diseases revealed no defects. A muscle biopsy and a blood sample were therefore taken at the age of 9 to further investigate the cause of his muscle problems. Blood samples were also obtained from his parents, siblings and the maternal relatives.

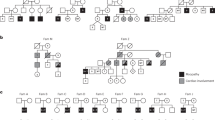
Detection, segregation and molecular consequence of the chromosomal duplication covering part of the MACF1 gene locus.
The family pedigree (a) shows segregation of the duplication on chromosome 1p34.4 (circles: women, squares: men, filled symbols: affected, open symbols: unaffected individuals, semi-filled symbol: the carrier). Array CGH (d) and SNP analysis (supplemental figure 1) identified the duplication, which was detected in affected family members only using MLPA (supplemental figure 2). MACF1 mRNA (b) and protein (c) was reduced compared to a control sample. A peptide competition assay confirmed the specificity of the MACF1 antibody (c, MACF1 + peptide). Analyzing actin, dysferlin and beta-dystroglycan protein levels validated the reduced expression of MACF1 protein in the proband compared to a control. Full-length versions of all western blots are presented in supplemental figure 4.
Genetic analysis
With array Comparative Genomic Hybridization (array CGH) analysis the only chromosomal abnormality detected was a single duplicated region on chromosome 1p34.3 (chr1: 39497393–39800239;HG19). The duplication detected in the proband covered a large part of the MACF1 gene affecting all 4 major MACF1 isoforms, however, few smaller isoforms located at the very end of the gene were not affected by the duplication (figure 1d). No other genes were present in the duplicated region. The same duplication was detected in the probands eldest sister and their mother and was later identified in the youngest sister (proband 2) as well using Single-Nucleotide Polymorphism array (SNP-array) analysis (supplemental figure 1).
According to the database of genomic variants, the duplication has not been found in the normal population (http://projects.tcag.ca/variation/).
Inheritance pattern and clinical presentation of the affected relatives
The inheritance pattern was determined using a Multiplex Ligation-dependent Probe Amplification (MLPA) analysis of the children, the parents and the maternal relatives (supplemental figure 2). The unaffected sibling, the father and the maternal relatives did not share the duplicated region (figure 1a, supplemental figure 2) and all affected children were heterozygous for the mutation. The duplication must therefore have arisen in the mother. Interestingly, the family members carrying the duplication all share the symptoms of the proband, but to varying degree. The mother and the eldest sister are less affected only displaying mild, hypotonic symptoms. However, the youngest sister (proband 2), now aged 2 years displayed severe hypotonic symptoms from birth, which manifested as diminished ability to suckle and weakness in the upper extremities, combined with general hypotonia. Routine laboratory tests and metabolic workup revealed no abnormalities in any of the affected relatives.
The molecular outcome of the duplication is a reduced MACF1 gene product
By comparing the muscle biopsy from the proband to control tissue we investigated how the duplicated region affected gene expression of MACF1. We detected reduced expression of MACF1 mRNA relative to the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (figure 1b). This translated to protein level as well, where MACF1 was markedly reduced relative to actin when comparing the proband sample to a control using western blotting (figure 1c). To validate the reduction of MACF1 protein we analyzed the expression of proteins related to other types of muscular dystrophy. Expression of dysferlin, the gene underlying Limb Girdle Muscular Dystrophy type 2B15, was equally expressed in proband and control samples. The same was observed for full-length β-dystroglycan (43DAG), a muscle membrane protein commonly affected by proteolysis in sarcoglycanopathy and Duchenne muscular dystrophy16.
Morphological analysis reveals discrete ultrastructural changes
Histologic and immunohistochemical analyses of the proband biopsy revealed no obvious skeletal muscle abnormalities (supplemental figure 3) and electron microscopy confirmed the normal gross appearance of the muscle ultrastructure in the proband (figure 2g) compared to a control (figure 2n) but also revealed discrete ultra structural changes in endothelial and satellite cells (figure 2a–f, m). In endothelial cells we could detect membrane foldings in tight junctions between endothelial cells, inside the endothelial cells and in membranes facing the muscle fiber (figure 2d and 2c). In the satellite cells, membrane foldings were mostly observable in the cell periphery and appeared as collapsed or folded structures (figure 2a and 2d). In a muscle biopsy from the youngest, most severely affected sister (proband 2) we found no obvious skeletal muscle abnormalities using histology and immunohistochemistry (data not shown). However, with electron microscopy we detected the same changes in satellite (figure 2h and 2i) and endothelial cells (figure 2j) as observed for the proband. Specifically, we observed the membrane changes at the cell periphery of satellite cells (figure 2i) and we observed changes in the tight junction membranes between endothelial cells (figure 2j). None of these membrane changes were observed in control muscle (figure 2o and 2p). Moreover, we detected cylindrical shaped electron dense membrane structures (figure 2k) and membranous whorls (figure 2l), a further indication of myopathy, since membranous whorls have been associated with e.g. inclusion body myositis17. These membranous whorl structures were very pronounced in proband 2 and based on these findings we re-examined the proband and detected similar whorl structures in this patient as well (figure 2m).

Normal muscle architecture with discrete ultrastructural changes due to reduced MACF1 gene product.
Transmission electron microscopic analyses of muscle biopsies from the proband (a–g) and his youngest sister (proband 2, h–l). In (a) a satellite cell from the proband is shown and the cell periphery having a severely affected membrane structure is magnified in (d, arrows). In (b) an endothelial cell from the proband is shown and this cell displays membrane foldings facing the myofiber (magnified in (c), arrows). In (e) and (f) another satellite cell from the proband is shown, again displaying an abnormal cell membrane structure that appears folded or whorled at the cell periphery (f, arrows). In (g) normal organization of myofibrils and z-bands in the proband is shown. In (h) and (i) a satellite cell presenting the same phenomenon in proband 2 as for the proband, with folded cell membrane structure at the periphery, is shown (i, arrows). In (j) an endothelial cell from proband 2 is shown. This endothelial cell displays a normal tight junction (j, white arrow) but also an abnormal, folded membrane structure at a tight junction (j, black arrows). An electron dense cylindrical membrane structure with membrane whorls at the periphery is observed in proband 2 (k, arrows). Proband 2 further displays large membranous whorl structures (l, arrows). These structures were also detected in the proband (m, arrows). In (n) normal ultrastructure from a control is shown. In (o) two satellite cells from control muscle are shown and in (p) an endothelial cell surrounding a vessel from a normal control is shown. SC = satellite cell, EC = endothelial cell, L = vessel lumen, M = membrane structure, TJ = tight junction, TJ* = abnormal tight junction, C = cylindrical membrane structure, MW = membranous whorl. Scale bars in a: 2000 nm; b: 5000 nm; c: 1000 nm; d: 2000 nm, e: 2000 nm, f: 1000 nm, g: 2000 nm; h: 1000 nm; i: 500 nm; j: 1000 nm; k: 1000 nm; l: 1000 nm; m: 500 nm; n: 2000 nm; o: 5000 nm; p: 5000 nm.
Impaired MACF1 function results in reduced motility
To address if the reduced motility and the internal structural changes observed in the proband and in proband 2 were caused by reduced MACF1 expression, we used RNA interference to knock down the vab-10 gene18 in C. elegans. The C. elegansvab-10 gene encodes multiple isoforms, which define two major isoform groups, VAB-10A and VAB-10B, which resembles human plectin and MACF1, respectively19. Since a null mutation of vab-10 (h1356) is lethal19, we knocked down the vab-10 locus by RNAi at the L3 larvae stage (app. 36 hours old) and analyzed the movement behavior after 24, 48 and 72 hours (figure 3a). Knock down of total vab-10 expression clearly decreased the ability to move compared to controls (24 h: p = 0.0087; 48 h: p < 0.0001) and after 72 h none of the RNAi-treated animals were capable of moving (p = 0.0001). However, the worms were still alive since they could move their heads slightly (see supplemental video 1 and 2). VAB-19 is a protein that is involved in attachment of the C. elegans body muscle to the epidermis and is a regulator of the actin cytoskeleton20. Knock down of vab-10 in transgenic animals expressing VAB-19::GFP clearly demonstrated that the attachment between the epidermis and the underlying muscles was defective while the overall tissue structure remained intact (figure 3b). Consistently, we also showed a similar phenotype by electron microscopic analyses (figure 3c).

Functional consequence of knocking down the vab-10 locus in Caenorhabditis Elegans.
Vab-10 knock down decreased the ability of C. elegans to move compared to controls (a, 24 h: p = 0.0087; 48 h: p < 0.0001) and after 72 h none of the RNAi-treated animals were capable of moving (p = 0.0001). We observed that knock down of vab-10 resulted in a collapse of the VAB-19 protein structure, which no longer localized to the muscle-epidermis interface20 (b, scale bar = 25 μm). Using transmission electron microscopy of vab-10 RNAi-treated worms and controls (c, scale bar = 1 μm) we confirmed an uncoupling between the epidermis and the underlying muscle tissue (indicated by double-pointed arrow in the vab-10 RNAi worm image, red arrows in the control worm image point towards the muscle-epidermal interface).
Discussion
Our investigations identified a patient with a novel type of myopathy characterized by periodic hypotonia, facial weakness, lax muscles, contractures and muscle pains. Our data suggested the cause to be a duplication on chromosome 1p34.4 resulting in reduction of the MACF1 gene product. Despite the overt symptoms very few changes were found in the muscle structure. This is however also observed in the myasthenic syndromes where the muscle biopsies usually show none or very few pathological changes. It could therefore be questioned whether the ultrastructural changes are myopathic in nature, however, we found that one of the more severely affected relatives of the patient, namely his youngest sister, displayed the same ultrastructural changes in the muscle as the patient, supporting a possible connection between the presence of the MACF1 duplication, the ultrastructural changes observed and the myopathic features.
The heterozygous phenotype of the affected individuals in this family might explain the manifestation of this disorder, since a complete knock down of this gene is known to be lethal8,19. This family therefore provides us with a unique insight into the mechanism of MACF1 in humans. Since MACF1 is ubiquitously expressed, it is intriguing that all affected subjects have developed normally and do not show signs of organ defects, especially considering the morphologically disturbed phenotypes observed in non-lethal C. elegansvab-10 mutants19. MACF1 heterozygous mice appear normal and fertile8, however, these mice have not ben analyzed for morphological changes in the skeletal muscles or muscle-related problems. In other mouse models of neuromuscular conditions such as Duchenne muscular dystrophy and dysferlinopathy milder phenotypes are also observed when comparing to the human condition21,22 and in these models, muscle-related problems are often only observable or comparable to the human disease under forced conditions e.g. treadmill exercise23 generally suggesting compensatory mechanism(s) or different pathological manifestations in mice compared to humans. In the proband as well as the other affected family members there are no cardiac disturbances even though MACF1 is expressed by cardiac tissue24 suggesting that the role of MACF1 in striated muscle differs between organ types. A recent study shows that MACF1 is dispensable for normal cardiac function but is important for the heart's ability to adapt to pressure overload25. Thus, this family must be monitored closely since the lack of cardiac involvement could relate to an inability of the family to undertake vigorous activity and hence subject the heart to sufficient strain. Cardiac involvement may occur later on as in other types of dystrophy e.g. Duchenne muscular dystrophy26.
MACF1 contains 23 dystrophin-like spectrin repeats and a plakin-like domain with homology to dystonin/BPAG1 and has been classified as a spectraplakin4,18. Interestingly, 3 of the major MACF1 isoforms contains an N-terminal actin-binding domain, whereas the major isoform 4 contains 8 plectin-repeats instead of the actin binding domain and these plectin domains are encoded by one exon, thus this MACF1 isoform has a similar genetic structure to plectin12. Therefore MACF1 can be considered a protein that possesses several distinct features related to cytoskeletal proteins. Dystonin/BPAG1 has been implicated in regulation of both z-lines and costameres in striated muscle27, mutations in dystrophin causes Duchenne Muscular Dystrophy characterized by membrane ruptures and severe muscle necrosis and inflammation28 and plectin deficiency causes muscular dystrophy associated with blistering of the skin29. The normal skeletal muscle architecture without necrosis in the proband suggests that the observed reduced expression of MACF1 has no effect on these structures and works through a different mechanism. However, it must be taken into consideration that these patients has an unaffected expression of few, very small MACF1 isoforms with currently unknown function, thus we do not know if or how these isoforms may affect the phenotype of the patients.
When observing the muscle ultrastructure we find subtle membrane related changes in endothelial cells and satellite cells in the proband (figure 2a–f). Specifically, the membranes appear folded and abnormal suggesting that lack of MACF1 impacts on normal membrane structure but without concomitant cellular necrosis. In the youngest sister (proband 2) we observe a pronounced presence of cylindrical membrane structures and membrane whorls (figure 2k and 2l), a known myopathic feature17, which after re-examination of the proband is also detected in this patient, suggesting that lack of MACF1 in addition to the cellular changes also results in classic myopathic changes. A lethal mutation in kakapo, the drosophila MACF1 homologue, results in detachments of the muscles from the cuticle due to impaired organization of the microtubules in the epidermal cells. However, the muscles remain attached to each other30. Moreover, it has recently been shown that MACF1 is recruited to the membrane in a complex fully dependent on the presence of microtubules31. The ultrastructural defects in the cellular membranes we observe could therefore be caused by insufficient microtubule stabilization in the satellite cells and endothelial cells. In other types of myopathies ultrastructural findings without pathological causation are a well-known phenomenon e.g. the undulating tubules associated with dermatomyositis32. Thus whether the ultrastructural changes are directly causing the pathology or is just an epi-phenomenon we cannot say.
Using C. elegans as a model we provide evidence that the impaired mobility is coupled to MACF1. Vab-10 RNAi greatly interfered with the worm's ability to move and after 72 h the worms treated with RNAi were incapable of movement, except for weak attempts to move their heads. Furthermore, vab-10 RNAi treated worms showed ultra-structural defects with an uncoupling between epidermis and the underlying muscle tissue, which is in line with a previous study19. The ultra-structural membrane disturbances resulting in this uncoupling between the cellular membrane and the underlying cytoskeleton does not disrupt the overall gross architecture of the skeletal muscle tissue, suggesting that the mechanism is associated with membrane attachment and not intracellular cytoskeletal structure formation. The Vab-10 gene encodes two isoforms, A and B, of which VAB-10A encodes the plectin homologue and VAB-10B is the MACF1 homologue18. It has been shown that it is VAB-10A that mediates the connection between the muscle and the cuticle thereby linking the muscle to the epidermis whereas VAB-10B is involved in control of epidermal thickness by linking the extracellular matrix to the epidermis19. Based on these observations it was suggested that each isoform protects the tissue from either internal (VAB-10B) or external force generation (VAB-10A)19. The RNAi construct used in the present study knocks down both isoforms and the predominant effects we observe are uncoupling of the muscle from the epidermis (figure 3c) as well as an inability for the worms to move around on the plates (supplemental videos 1 and 2). The effect on the muscle-epidermis-cuticle connection is therefore in coherence with the previous findings for both VAB-10A and VAB-10B19. However, the locomotor phenotype has not previously been shown in adolescent animals. Although the present data do not allow us to distinguish their specific roles in locomotion, we interpret that these movement defects may be associated with simultaneous loss of both isoforms. Since the vab-10 locus encodes the only spectraplakin in C. elegans the findings in this model cannot be transferred directly to the human condition. Nevertheless, since C. elegans is incapable of moving after reducing total VAB-10 protein expression we suggest that the reduced protein expression of MACF1 in the patient may be the underlying cause of the generalised muscle weakness and hypotonia especially considering that one of the affected major isoforms share structural similarities to plectin as well12. The reduced expression of MACF1 has only been determined for at muscle biopsy thus we cannot rule out that other tissues might be directly affected such as tendons. We foresee that the present work lays an excellent foundation to further delineate the functions of MACF1 in metazoans as well as humans.
Methods
Ethical statement
Informed consent was obtained from all human subjects and the data obtained using blood samples and biopsies in this study has been laid before the Regional Scientific Ethical Committee of Southern Denmark, which has approved that the data can be published as a case report.
Array CGH
The Oxford Gene Technology Syndrome Plus v.2 format was used for array-CGH analysis. Labelling and hybridization were done according to standard protocol from Oxford Gene Technology and scanning was performed on a Perkin Elmer Scanarray gX scanner at a resolution of 5 μm. Genepix Pro 6.1 was used for feature extraction and Cytosure Interpret software v3.4.3 was used for data analysis.
MLPA
A custom MLPA probe was designed with the assistance of the online H-MAPD MLPA custom probe designer software (http://bioinform.arcan.stonybrook.edu/mlpa2/cgi-bin/mlpa.cgi) and designed to bind within the duplicated region on chromosome 1p34.3 (Specifically at bp position: 1:39,488,298–39,488,364; HG18). Custom probes were added to a P096 MR2 MLPA KIT from MRC Holland according to standard protocol.
SNP-array
The Illumina HumanCytoSNP-12 v2.1 beadchip format was used for SNP-array analysis. Labeling and hybridization were done according to standard protocol from Illumina and scanning was done on an Illumina HiScanSQ scanner. Illumina Genomestudio v2011.1 was used for quality control and Illumina Karyostudio v1.4.3 was used for data analysis.
RT-PCR
Total RNA was extracted using Trizol according to the manufacturers description (Life technologies. cat. # 15596026) and transcribed into complementary DNA (cDNA) using the High-Archive cDNA kit (Applied Biosystems, cat. # 4368813) according to the manufacturers description. 1 μl template of cDNA from the proband's sample and from a control sample was used to amplify a constant part of MACF112 (NM_012090.4) (Fwd primer located at the exon 81-82 boundary: 5′ CTGTGGAGCGGCAGCACAAGT 3′; Rev primer located at the exon 87–88 boundary: 5′ CTGCCTCCGCTGCGGGATTT 3′) and the endogenous control GAPDH (Fwd primer: 5′ AAGGCTGGGGCTCATTTGCA 3′; Rev primer: 5′ GTGGTCATGAGTCCTTCCAC 3′) using LA Taq (Takara, cat.# RR002A). The MACF1 mRNA product is 949 bp and the GAPDH mRNA product is 212 bp. Cycling conditions: 95°C for 2 min followed by 30 cycles: 95°C 20 sec, 62°C 20 sec, 72°C 20 sec then 72°C for 7 min and 4°C hold. PCR products were gel-extracted and sequenced to verify specificity of the amplification products.
Western blotting
Total protein was extracted from the proband and a control sample and loaded on a 3–8% Tris-Acetate gel without prior heating of the samples, run using Tris-Acetate running buffer for 1 h at 150 V constant and transferred to a PVDF membrane (0.45 μm pore size) using electro-blotting for 2 h at 200 V constant, with stirring and cooling. The membrane was blocked for 1 h at room temperature with 5% skimmed milk/TBS-T and incubated with primary antibodies over night at 4°C, shaking (mouse-anti-MACF133 (H00023499-A01, Novus Biologicals) 1:50, mouse-anti-beta dystroglycan, clone 43DAG (Novocastra) 1:100, mouse-anti-dysferlin, NCL-Hamlet (Novocastra) 1:100, mouse-anti-dystrophin, clone DY8 (Novocastra) 1:20). The following day blots were washed using TBS-T and incubated with goat-anti-mouse- HRP (DAKO) 1:1000 for 1 h at room temperature and developed using Enhanced Chemiluminescence kit (Invitrogen) and standard x-ray film. To verify the specificity of MACF1, a competition assay was performed were the MACF1 peptide used to produce the antibody (H00023499-Q01, Novus Biologicals, this peptide corresponds to aa in position 7121-7215 of 7388) was added to the MACF1 primary antibody before addition to the membrane (peptide-mix: 20 μl antibody +100 μl peptide +80 μl blocking solution was incubated for 1 h at room temperature, then 800 μl blocking solution was added and the solution used to probe the membrane).
Immunohistochemistry/histology
HE, Gomori trichrome and NADH staining were performed according to standard protocols. Immunohistochemistry was performed on frozen unfixed tissue for CD56 (Cell Marque, 156R) 1:500, MHC slow (Novocastra, NCL-MHCs) 1:50, MHCfast (Sigma, M4276) 1:8000, MHC neonatal (Novocastra, NCL-MHCn) 1:10, dystrophin rod (Chemicon, MAB1692) 1:10, dystrophin C-terminus (Chemicon, MAB 1694) 1:5, dystrophin N-terminus (Novocastra, NCL-DYSB) 1:20 using BenchMark Ultra (Roche) according to standard protocol. Incubation time for primary antibodies: 32 min at 36°C. Detection system used: OptiView-DAB (Roche). CD68, EBMII (Dako) was stained on acetone fixed (10 min) tissue.
Electron microscopy
Muscle tissue from the proband, a human muscle control sample, C.elegansvab-10 RNAi and control worms were fixated in 2% glutaraldehyde/PBS, stained with 1% osmiumtetraoxide/PBS, dehydrated and embedded in epon (epoxyresin). Ultrathin sections (60 nm) were cut on a Leica Ultracut UCT and placed on a grid, contrasted using 3% uranylacetate/H2O and leadcitrate (Leica Ultrostain 2) and analyzed with transmission electron microscopy on a Philips EM 208 electron microscope.
Culturing and handling of C. elegans and E. coli strains
Worms were cultured using standard protocols on plates seeded with different E. coli strains including OP50 and HT115 for RNAi34. Wild-type N2 Bristol and the transgenic strain CZ3103 expressing a translational fusion between VAB-19 and GFP (juIs167 [VAB-19::GFP + pRF4]) (described in20) were obtained from the Caenorhabditis elegans Genetics Center. For RNAi studies we used an RNAi vab-10 clone (ZK1151.1) from the Ahringer RNAi library as described previously35. The RNAi clone was confirmed by sequencing. RNAi by feeding was performed according to Ahringer36. HT115 bacteria transformed with the empty vector L4440 was used as a negative control. Synchronized L3 worms grown at 20°C were picked to freshly made RNAi plates and assayed after 24, 48 and 72 hours. For epifluorescence microscopy worms were mounted on freshly made 2% agarose pads and anaesthetized by 10 mM levamisole (Sigma) in M9 and VAB-19::GFP was visualized at 200× and 1000× using a Leica DMI 6000 B microscope. All images at a specific magnification were acquired using identical settings and exposure time. Videos were recorded after 24, 48 and 72 hours of vab-10 RNAi using a Leica MZ6 microscope equipped with an Infinity2 video camera (Lumenera, Ottawa, Canada). The videos were analyzed for worm movement behavior using ImageJ.
Statistical analysis
Statistical analysis of worm movement behavior was performed as a two-tailed Mann Whitney test with Gaussian Approximation since the data sets were not normally distributed. Distribution within each data set was tested using D'Agostino & Pearson normality test. All statistical analyses were carried out using GraphPad Prism 5 and with an alpha value of 0.05.
References
Doherty, G. J. & McMahon, H. T. Mediation, modulation and consequences of membrane-cytoskeleton interactions. Annu Rev Biophys 37, 65–95 (2008).
Sonnenberg, A. & Liem, R. K. Plakins in development and disease. Exp Cell Res 313, 2189–203 (2007).
Sun, Y. et al. Molecular cloning and characterization of human trabeculin-alpha, a giant protein defining a new family of actin-binding proteins. J Biol Chem 274, 33522–30 (1999).
Leung, C. L., Sun, D., Zheng, M., Knowles, D. R. & Liem, R. K. Microtubule actin cross-linking factor (MACF): a hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J Cell Biol 147, 1275–86 (1999).
Kodama, A., Karakesisoglou, I., Wong, E., Vaezi, A. & Fuchs, E. ACF7: an essential integrator of microtubule dynamics. Cell 115, 343–54 (2003).
Wu, X., Kodama, A. & Fuchs, E. ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell 135, 137–48 (2008).
Wu, X. et al. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3beta. Cell 144, 341–52 (2011).
Chen, H. J. et al. The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev 20, 1933–45 (2006).
Lewis, S. L. et al. Dkk1 and Wnt3 interact to control head morphogenesis in the mouse. Development 135, 1791–801 (2008).
Gupta, T. et al. Microtubule actin crosslinking factor 1 regulates the Balbiani body and animal-vegetal polarity of the zebrafish oocyte. PLoS Genet 6, e1001073 (2010).
Bouameur, J. E., Favre, B. & Borradori, L. Plakins, a Versatile Family of Cytolinkers: Roles in Skin Integrity and in Human Diseases. J Invest Dermatol (2013).
Gong, T. W., Besirli, C. G. & Lomax, M. I. MACF1 gene structure: a hybrid of plectin and dystrophin. Mamm Genome 12, 852–61 (2001).
Koenig, M. et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–17 (1987).
Guo, L. et al. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 81, 233–43 (1995).
Liu, J. et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 20, 31–6 (1998).
Matsumura, K. et al. Proteolysis of beta-dystroglycan in muscular diseases. Neuromuscul Disord 15, 336–41 (2005).
Carpenter, S. & Karpati, G. The pathological diagnosis of specific inflammatory myopathies. Brain Pathol 2, 13–9 (1992).
Roper, K., Gregory, S. L. & Brown, N. H. The ‘spectraplakins’: cytoskeletal giants with characteristics of both spectrin and plakin families. J Cell Sci 115, 4215–25 (2002).
Bosher, J. M. et al. The Caenorhabditis elegans vab-10 spectraplakin isoforms protect the epidermis against internal and external forces. J Cell Biol 161, 757–68 (2003).
Ding, M., Goncharov, A., Jin, Y. & Chisholm, A. D. C. elegans ankyrin repeat protein VAB-19 is a component of epidermal attachment structures and is essential for epidermal morphogenesis. Development 130, 5791–801 (2003).
Sicinski, P. et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244, 1578–80 (1989).
Chiu, Y. H. et al. Attenuated muscle regeneration is a key factor in dysferlin-deficient muscular dystrophy. Hum Mol Genet 18, 1976–89 (2009).
Radley-Crabb, H. et al. A single 30 min treadmill exercise session is suitable for ‘proof-of concept studies’ in adult mdx mice: a comparison of the early consequences of two different treadmill protocols. Neuromuscul Disord 22, 170–82 (2012).
Bernier, G. et al. Acf7 (MACF) is an actin and microtubule linker protein whose expression predominates in neural, muscle and lung development. Dev Dyn 219, 216–25 (2000).
Fassett, J. T. et al. Microtubule Actin Cross-linking Factor 1 regulates cardiomyocyte microtubule distribution and adaptation to hemodynamic overload. PLoS One 8, e73887 (2013).
Spurney, C. F. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve 44, 8–19 (2011).
Dalpe, G. et al. Dystonin-deficient mice exhibit an intrinsic muscle weakness and an instability of skeletal muscle cytoarchitecture. Dev Biol 210, 367–80 (1999).
Hoffman, E. P., Brown, R. H., Jr & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–28 (1987).
Smith, F. J. et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet 13, 450–7 (1996).
Prokop, A., Uhler, J., Roote, J. & Bate, M. The kakapo mutation affects terminal arborization and central dendritic sprouting of Drosophila motorneurons. J Cell Biol 143, 1283–94 (1998).
Margaron, Y., Fradet, N. & Cote, J. F. ELMO recruits actin cross-linking family 7 (ACF7) at the cell membrane for microtubule capture and stabilization of cellular protrusions. J Biol Chem 288, 1184–99 (2013).
Goebel, H. H. & Stenzel, W. Ultrastructural myopathology in the molecular era. Ultrastruct Pathol 37, 328–31 (2013).
Khositseth, S. et al. Quantitative protein and mRNA profiling shows selective post-transcriptional control of protein expression by vasopressin in kidney cells. Mol Cell Proteomics 10, M110 004036 (2011).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Kamath, R. S. et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–7 (2003).
Kamath, R. S. & Ahringer, J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–21 (2003).
Acknowledgements
Supported by a grant from The Danish Council for Independent Research and a grant from the Villum Foundation to L.H.J. We thank Kirsten Hansen for technical assistance with electron microscopy and the patients and family for their support.
Author information
Authors and Affiliations
Contributions
L.H.J., M.M. and J.G. performed experiments and analyzed data, H.D.S. and S.V.J. provided human samples and patient data, N.J.F. provided the C. elegans models, L.H.J. and H.D.S. designed the study and L.H.J. wrote the paper. All authors discussed results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Supplemental video 1
Supplementary Information
Supplemental video 2
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Jørgensen, L., Mosbech, MB., Færgeman, N. et al. Duplication in the Microtubule-Actin Cross-linking Factor 1 gene causes a novel neuromuscular condition. Sci Rep 4, 5180 (2014). https://doi.org/10.1038/srep05180
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05180
This article is cited by
-
Replication of MACF1 gene variant rs2296172 with type 2 diabetes susceptibility in the Bania population group of Punjab, India
International Journal of Diabetes in Developing Countries (2018)
-
Genetic Variants of Microtubule Actin Cross-linking Factor 1 (MACF1) Confer Risk for Parkinson’s Disease
Molecular Neurobiology (2017)
-
Microtubule plus-end tracking proteins in neuronal development
Cellular and Molecular Life Sciences (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
