
Abstract
Sinonasal squamous cell carcinomas (SCC) are rare tumors, etiologically related to occupational exposure to wood and leather dust. In spite of surgical and radiotherapeutic advances, the 5 year survival is still 30–50%. Therefore, alternative treatment options are needed. We report the establishment and characterization of six unique human sinonasal SCC cell lines, named SCCNC1, 2, 4, 5, 6 and 7. In vitro growth and invasion characteristics were evaluated and genetic profiles were compared to those of the original primary tumors. The population doubling times ranged from 21 to 34 hours. Cell lines SCCNC2 and 7 were highly invasive in matrigel. Five cell lines carried a high number of copy number alterations, including amplifications and homozygous deletions, while one showed only three abnormalities. Sequence analysis revealed three cell lines with TP53 mutation and none with KRAS or BRAF. Overexpression of p53 was observed in five and of EGFR in four cell lines. None of the cell lines showed strong immunopositivity of p16 or presence of human papilloma virus. In conclusion, we have created six new cell lines that are clinically and genetically representative of sinonasal SCC and that will be a useful tool for the preclinical testing of new therapeutic agents.
Similar content being viewed by others
Introduction
Sinonasal squamous cell carcinomas (SCC) are malignant epithelial tumors that originate in the respiratory epithelium of the sinonasal cavities, respresenting about 3–6% of all head and neck cancers. The incidence is <1 case per 100,000 inhabitants per year, occurring predominantly among men with a mean age of presentation of 50 to 60 years1,2. Other, less frequent epithelial tumors in this anatomical region include intestinal-type adenocarcinoma, undifferentiated carcinoma, neuroendocrine carcinoma and neuroestesioblastoma3. Sinonasal SCC arise predominantly in the maxillary sinus and the nasal cavity and approximately 30% of cases are etiologically related to professional exposure to textile, leather, wood or aluminium4,5. In contrast to most head and neck cancers, tobacco does not appear to play a key role. However, there is evidence for a 2–3 fold increase in sinonasal SCC risk from tobacco smoking6,7. Finally, human papilloma virus (HPV) types 16 and 18 may be implicated in the development of sinonasal SCC, mainly in those cases with malignant transformation of inverted papillomas which occurs in 5 to 15% cases8,9.
Sinonasal SCC are often diagnosed at advanced stages due to the insidious symptoms. Distant and lymph node metastasis are exceptional (5–15%). Advanced T stage, base skull involvement, orbital extension and local recurrence are indicators of poor survival. Standard therapeutic modalities include surgery followed by radiotherapy, sometimes with adjuvant chemotherapy10. The principal cause of mortality is local recurrence (40–50% of all cases), occurring even when the tumor was extracted with wide tumor-free resection margins. The overall 5-year survival is 30–50%1,2,7. Therefore, additional options for treatment are needed for the management of sinonasal SCC.
Our knowledge on the genomics of cancer, including sinonasal SCC, is increasing and allows a more refined tumor classification and more personalized therapeutic opportunities based on the genomic profiles. Stable tumor cell lines as a model system retaining the biological properties of primary cancer cells are an important tool for the development and testing of such new therapeutic agents. Here, we report the establishment of six new human sinonasal SCC cell lines, characterized by their morphology, population doubling time, invasiveness in matrigel and genetic aberrations. In addition, we demonstrate that the cell lines are isogenic to and share most of the copy number alterations with the original primary tumors from which they were derived.
Methods
Patient histories and tumor characteristics
All cell lines were derived from fresh samples of previously untreated, primary SCC originating in the maxillary sinus. Informed consent was obtained from all patients. Surgical tissue specimens were collected and experiments were performed in accordance with the approved guidelines of the Ethics Committee of the Hospital Universitario Central de Asturias. None of the six patients had a history of exposure to wood, leather or textile dust, or industrial chemical substances as glues, formaldehyde, chrome, or nickel. The clinical staging of sinonasal tumors is based on location and extent, according to the AJCC 7th edition11. An overview of patient and tumor characteristics is given in table 1.
SCCNC1
A 66-year-old male was diagnosed and treated for a T2N1M0 moderately differentiated SCC of the left maxillary sinus. A craniofacial resection was performed and the tumor was resected in monobloc with negative margins. Moreover, an ipsilateral modified radical neck dissection was performed. After surgery, the patient received 66 Gy of radiotherapy. After 6 months, the patient developed a local recurrence and was treated with surgery concomitant chemoradiotherapy. The patient subsequently developed another local recurrence and died with local disease at 16 months.
SCCNC2
A 71-year-old male was diagnosed and treated for a T4N1M0 moderately differentiated SCC of the left maxillary sinus. The patient was primarily treated with surgery through lateral rhinotomy, however, because of positive surgical margins the patient received postoperative chemoradiotherapy. The patient developed a recurrence at the medial canthus of the left eye and in the ipsilateral neck 7 months after treatment and underwent orbital exenteration and left modified radical neck dissection. The patient developed hematogenous metastases and died at 11 months after diagnosis.
SCCNC4
A 73-year-old female was diagnosed and treated for a T2N1M0 poorly differentiated SCC of the right maxillary sinus. Previously in another hospital the patient was diagnosed and treated for inverted papilloma in the right nasal cavity. The patient underwent right medial maxillectomy and right modified radical neck dissection and subsequently received postoperative radiotherapy. A local recurrence developed 5 months later and the patient was treated by total maxillectomy. However, after 10 months the patient had an unresectable local recurrence and died at 22 months after diagnosis.
SCCNC5
A 79-year-old male was diagnosed and treated for a T2N0M0 well differentiated SCC of the right maxillary sinus. He received surgical management through an infratemporal maxillectomy with negative margins and free flap reconstruction. The patient developed a neurological complication in the early postoperative period and died due to this complication.
SCCNC6
A 63-year-old male was diagnosed and treated for a T2N0M0 well differentiated SCC of the left maxillary sinus. The tumor was surgically removed by left infratemporal maxillectomy with negative margins and the patient received postoperative radiotherapy. A bilateral neck recurrence developed 21 months after treatment and the patient was treated by bilateral modified radical neck dissection. He is currently alive and disease-free at 32 months.
SCCNC7
A 48-year-old male was diagnosed and treated for a T4aN2bM0 poorly differentiated SCC of the right maxillary sinus. He underwent surgical management through a total maxillectomy and right modified radical neck dissection and received postoperative radiotherapy. After 3 months, the patient developed an unresectable locoregional recurrence and was treated with palliative chemotherapy. Shortly after he died with disease at the primary site.
Cell line establishment
Fresh tumor samples from the operating theatre were cut into several small fragments, transferred to dry 25 cm2 culture flasks and covered with a drop of MEM culture medium, supplemented with 10% FBS, 100 U/ml penicillin, 200 μg/ml streptomycin, 2 mM L-Glutamine and non-essential amino acids 100 μM (PAA Laboratories GmbH, Pasching, Austria) and incubated in 5% C02 at 37°C. Initial outgrowth of both tumor and fibroblast cells was observed after 7 days. Possible overgrowth by fibroblasts was prevented by repeated selective trypsinizations. At the moment of writing this manuscript, all cell lines have been in culture for over 24 months and have been passaged more than 80 times. Possible contamination by Mycoplasma was checked using the LONZA MycoAlert Mycoplasma Detection Kit (LONZA, Rockland CE, USA).
Proliferation and in vitro invasion
The growth rate was assessed by seeding 500,000 cells in 25 cm2 culture flasks and counting at 24–48–72 hour intervals using a hemacytometer. Cellular invasion was evaluated by the ability of cells to invade matrigel membranes (BioCoat Matrigel Invasion Chambers, BD Biosciences, San José CA, USA). Cells cultured in complete MEM medium were trypsinized, washed two times in PBS and the taken up in MEM medium without supplements. A suspension containing 20,000 cells were placed in the upper compartement and the lower compartement was filled with complete MEM medium. Cells were incubated for 24 hours in 5% C02 at 37°C. Subsequently, non-invading cells on the membranes were washed off with PBS and the membranes were stained with 0.5% cristal violet in methanol. The number of invading cells were counted using a light microscope with a 10× objective. Short-term cultured keratinocyte cells derived from normal nasal mucosa and grown in a serum-free human keratinocyte growth medium (Keratinocyte SFM, Invitrogen, Barcelona, Spain) were use as negative control. Both assays were performed in triplicate.
Immunohistochemistry and immunofluorescence
The following antibodies were used: anti-p53 clone DO-7 (DAKO, Glostrup, Denmark), anti-p16 clone E6H4 (CINTEC, MTM Laboratories, Madrid, Spain) and anti-EGFR clone 2-18C9 (DAKO, Glostrup, Denmark). Positive and negative control samples (cell line spin downs) for EGFR were provided by the the manufacturer. A HPV-positive cervix carcinoma sample was used as a positive control for p16 and a TP53 mutated sinonasal intestinal-type adenocarcinoma sample for p5312,13. Immunohistochemistry was performed using an automatic staining workstation (Dako Autostainer, Dako Cytomation, Glostrup, Denmark) with the Envision system and diaminobenzidine chromogen as substrate. Immunofluorescence took place on SCCNC cells grown on microscope slides. Results were visualized by fluorescent secondary antibodies (Goat-anti-Rabbit, Alexa 488 and Goat-anti-Mouse, Alexa 488 (InVitrogen Molecular Probes, Barcelona, Spain) and DAPI counterstaining (Vector Laboratories, Burlingame CA, USA) and evaluated using the Olympus BX-61 fluorescence microscope. The immunostainings were evaluated by two independent observers (CGI and MH). P16 immunostaining was scored as negative (0), weak to moderate staining (1+) or moderate to strong diffuse cytoplasmic and nuclear staining (2+). P53 immunostainings was evaluated as positive when >10% of the malignant cells showed nuclear staining and EGFR immunostaining when strong membranous and/or cytoplasmic staining was observed in at least 10% of tumor cells.
DNA extraction and short tandem repeat (STR) genotyping
Tumor DNA was extracted with Qiagen extraction kits (Qiagen GmbH, Hilden, Germany) from the primary tumor, the cell line and from peripheral blood lymphocytes of the same patient. DNA from the cell lines was obtained at passages bewteen 6 and 10. STR genotyping was performed on DNA from the cell lines and from peripheral blood lymphocytes using the Promega Powerplex 16 system (Promega Biotech Ibérica SL, Barcelona, Spain) to amplify fifteen STR loci (Penta E, D18S51, D21S11, TH01, D3S1358, FGA, TPOX, D8S1179, vWA, Penta D, CSF1PO, D16S539, D7S820, D13S317 and D5S818) and Amelogenin by PCR.
TP53, BRAF and KRAS mutation analysis
TP53 mutations in exons 5–9, KRAS in exon 2 (codons 12 and 13) and BRAF exon 15 (V600E) were analyzed by direct sequencing using the ABI PRISM 3100 and 3730 Genetic Analyzer, (Applied Biosystems, Foster City CA, USA) as described previously13,14 using the following primers:
TP53 Exon 5–6: Forward TGACTTTCAACTCTGTCTCC Reverse GCCACTGACAACCACCCTTA
TP53 Exon 7: Forward CCAAGGCGCACTGGCCTCATC Reverse TCAGCGGCAAGCAGAGGCT
TP53 Exon 8–9: Forward GACCTGATTTCCTTACTGCCTC Reverse GACTGGAAACTTTCCACTTGA
KRAS Exon 2: Forward TACTGGTGGAGTATTTGATAGTG Reverse CTGTATCAAAGAATGGTCCTG
BRAF Exon 15: Forward CTTCATAATGCTTGCTCTGATAGG Reverse GCATCTCAGGGCCAAAAAT
Sense and antisense sequencing was performed for confirmation.
HPV DNA detection
The quality of the extracted DNA was checked by PCR amplification of β-globin (forward primer 5′-ACACAACTTGTGTGTTCACTAGC-3′ and reverse primer 5′-CAAACTTCATCCACGTTCACC-3′). PCR with MY11/GP6+ primers (site-directed L1 fragment of HPV) was performed in order to detect a broad spectrum of HPV genotypes15. Briefly, the PCR was performed in 25 μl of reaction mixture containing 1× PCR buffer, 2 mmol/L MgCl2, 50 μmol/L of each deoxynucleoside, 0.5 μmol/L of sense and antisense primers, 10 μl of DNA sample and 1 U Taq DNA polymerase (Promega Biotech Iberica S.L. Madrid, Spain), by thermal profile of 35 cycles: denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec and extension at 72°C for 1 min, with an initial denaturation at 94°C for 5 min and a final extension at 72°C for 10 min. The amplified DNA fragments of approximately 200 bp were identified by electrophoresis in 1.5% agarose gel with ethidium bromide.
Microarray CGH
Microarray CGH analysis was performed as described previously, using a 180 k oligonucleotide array (SurePrint G3 Human CGH Microarray Kit 4 × 180 K, Agilent Technologies, Palo Alto CA, USA)16. Images were acquired using a Microarray scanner G2505B (Agilent Technologies, Palo Alto CA, USA). Analysis and data extraction were quantified using feature extraction software (version 9.1, Agilent Technologies, Palo Alto CA, USA). Gains and losses were defined as deviations of 0.2 or more from log2 ratio = 0.0. High-level amplification was considered when at least 2 neighboring clones reached a log2 ratio of >2.0 and homozygous deletions (HDs) were defined as two or more consecutive oligonucleotides with a log2 ratio of <−3. The possibility of copy number variations (rather than copy number alterations) was excluded by using normal DNA of the same patient as reference. The microarray CGH data are accessible through GEO Series accession number GSE57201.
Results
Cell morphology
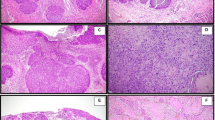
SCCNC1, SCCNC2, SCCNC4, SCCNC5 and SCCNC6 grew as a tightly packed monolayer of small polygonal shaped cells, similar to the morphology that can be seen in the H&E sections of the patient's primary tumors. SCCNC7 grew as a monolayer of fusiform shaped cells (Figure 1). The H&E images demonstrate the differences in histological grade among the primary tumors, SCCNC5 and SCCNC6 being well, SCCNC1 and SCCNC2 moderately and SCCNC4 and SCCNC6 poorly differentiated (Figure 1).

Photomicrographs of the in vitro growing cell lines (left column) and representative H&E stained paraffin sections of the corresponding primary tumors (right column).
Proliferation and invasion
The cell lines showed great differences in plating efficiency after trypsinization, SCCNC7 and SCCNC5 settled and continued proliferating quickly, whereas the other cell lines were slower in adhering and resuming proliferation. In the exponential growth phase, all cell lines showed population doubling times between 21 and 27 hours, except for SCCNC6 which demonstrated a doubling approximately every 34 hours (Figure 2A). Mycoplasma was absent in all cell lines. Using the matrigel invasion assay with normal keratinocytes cells as experimental control, we found none of the originally 20,000 seeded cells in the matrix. Cell lines SCCNC2 and SCCNC7 showed a strong capacity to invade with 439 and 153 cells in the matrix after 24 hours, respectively. SCCNC1 showed a moderate invasiveness with 81 cells, while SCCNC4, SCCNC5 and SCCNC6 hardly showed any invasion with less than 25 cells (Figures 2B and 2C).

Proliferation and invasion in matrigel.
(A) In the exponential growth phase, the cell lines showed population doubling times between 21 and 34 hours. After passaging, SCCNC7 and SCCNC5 settled and continued proliferating quickly, whereas the other cell lines were slower in adhering and resuming proliferation. (B and C) Cell lines SCCNC2 and SCCNC7 showed the strongest capacity to invade with 439 and 153 cells in the matrix after 24 hours, respectively. All experiments were done in triplicate.
Genetic identity and DNA copy number profile
Short tandem repeat fingerprinting performed on DNA from the cell lines and blood lymphocytes from the patients confirmed the individual identity of all cell lines. All cell lines showed at least 90% identical short tandem repeats. Moreover, microarray CGH analysis of DNA extracted from frozen tumor tissue of the primary tumor from which the cell lines were derived, showed an identical pattern of copy number alterations (Figure 3), although the amplitude of the observed gains and losses was higher in the cell lines.

Microarray CGH profiles of cell lines at passages between 6 and 10 (left column) and their corresponding primary tumors (right column).
All data points are expressed as log2-ratios, ordered continuously from left to right as chromosome 1 up to chromosome X (here numbered as 23). In cell lines SCCNC1,2,4,6 and 7 many chromosomes show copy number alteration, including homozygous deletion and high level amplification. The majority of the aberrations are also present in the primary tumor. Cell line SCCNC5 harboured only three copy number changes and these were not detected in the primary tumor.
All cell lines except SCCNC5 showed complex, aneuploid karyotypes with multiple numerical and structural aberrations involving all chromosomes. SCCNC5 was diploid and harboured only three chromosomal abnormalities, a deletion at 2q, a gain at 5q and a whole chromosome 20 gain. Frequent losses were found at chromosomal band 8p, 13q (5 cell lines), 3p, 4q, 10p, 18q and 21q (4 cell lines). Recurrent gains were observed at 20p, 20q (all cell lines), 3q, 7p, 8q, 11q, 14q (5 cell lines) and 18p (4 cell lines). Amplification occurring in more than one cell line was found at 11q13 (4 cell lines), with the smallest region of overlap at position 69.093.227–70.110.393 bp, which include the genes CCND1, ORAOV1, FGF19, FGF4, FGF3, AND1 and FADD (Table 2). Other high level amplifications included 3q25, 7q21, 11p13, 11q12, 11q14, 13q21, 17q11, 18q11 and Xp11. Homozygous deletions indicating complete absence of DNA material were present only in cell lines SCCNC2 and SCCNC6 and are listed in Table 2.
TP53, KRAS, BRAF, EGFR, CDKN2A and HPV
Sequencing of TP53 exons 5–9 revealed missense mutations in three cell lines. SCCNC1 and SCCN7 both carried a transition c.844C > T (p.R282W) in exon 8 and SCCNC5 showed a transition c.511G > A (p.E171K) in exon 5. These three cell lines also showed a strong nuclear p53 protein overexpression in the primary tumor and in the cell line. Two additional cell lines showed p53 overexpression in absence of TP53 mutation (Figure 4). EGFR gene copy number gain (all cell lines except SCCNC5) did not always associate with EGFR protein overexpression, which was observed in cell lines SCCNC1, SCCNC4, SCCNC5 and SCCNC7, but not in SCCNC2 and SCCNC6 (Figure 5). No mutations were found in KRAS exon 2 (codons 12 and 13) and BRAF exon 15 (V600E). P16 expression was absent in all cell lines, while among the primary tumors only SCCNC4 showed a weak positivity (Figure 5). HPV analysis was negative for all cell lines and primary tumors.

P53 expression analysis by immunofluorescence on the in vitro growing cell lines (left column) and by immunohistochemistry on the corresponding primary tumors (right column), showing positivity in all cell lines except SCCNC4.

P16 (left column) and EGFR (right column) expression analysis by immunohistochemistry on the primary tumors.
SCCNC4 shows a weak p16 immunostaining. EGFR overexpression is observed in SCCNC1, 4, 5 and 7.
Discussion
Many different types of tumors arise in the sinonasal cavities. Their incidence is low, a fact that has hampered molecular-genetic studies on tumorigenic pathways and the testing of alternative treatment strategies. In vitro tumor cell lines are valuable tools for functional studies on processes as proliferation, differentiation, invasion and metastasis, as well as preclinical testing of new therapeutic agents.
We have performed an extensive literature search and found a total of 28 cell lines derived from 19 different sinonasal tumors (in four cases, two or more sister cell lines were derived from the same patient). Six of these 19 concern tumors of a different histological type than sinonasal SCC16,17,18,19,20 and three are in fact not derived of sinonasal respiratory mucosa, but of skin (nasal vestibule and septum)21,22,23,24,25 or the alveolar ridge26. Of the remaining ten ‘true’ sinonasal SCC cell lines, six are established from recurrent tumors or lymph node metastases22,23,24,27,28,29,30,31, leaving only four cell lines from primary, previously untreated tumors (Table 3)22,27,32,33,34. In the present paper we describe six new sinonasal SCC cell lines, all derived from previously untreated, primary tumors.
The clinical variety that is generally seen in sinonasal SCC is well represented by our six new cell lines: two cases are derived from T2N0, two T2N1 and two T4N1,2 tumors, two cases each showed well, moderate and poor differentiation and finally two patients were smokers and four non-smokers. While all cell lines grew well in vitro, their capacity to invade matrigel varied considerably: SCCNC2 and SCCNC7 showed a very high level of invasion, SCCNC1 intermediate and SCCNC4, SCCNC5 and SCCNC6 were not able to invade into the matrigel.
The genome-wide profile of gains and losses of the cell lines was similar to what has been described in primary sinonasal SCC35, including losses at chromosomal bands 3p, 4q, 8p, 10p, 13q, 18q and 21q, gains at 3q, 7p, 8q, 11q, 14q, 18p and 20q and amplification at 11q13. Most of these aberrations are also recurrent in head and neck SCC. A curious exception are gains at chromosome 5p and loss at 5q, these are very frequent in head and neck SCC but not so in sinonasal SCC35. Aside from recurrent genetic changes, we found several focal aberrations in the cell lines, both high level amplifications and homozygous deletions. Their exact size and localization are listed in table 3, together with the genes (if any) included in the focal aberration. Some of the homozygous deletions actually indicated the complete absence of DNA material. The possibility that they might be copy number variations (i.e. polymorphisms) was ruled out because we used normal DNA from the same patient as reference DNA. Focal aberrations are important because usually they concern very few genes and may indicate aberrant cellular pathways that could be useful targets for new anticancer drugs36. Amplification of chromosomal region 11q13, observed in four of the six cell lines, is a frequent event in sinonasal SCC35 as it is in head and neck SCC37,38 and other tumors39. Apart from reports on the prognostic significance of this alteration37,38,39, recent studies have indicated a role for amplification of CTTN and overexpression of cortactin (coded by CTTN) in the progression from premalignant lesions to invasive carcinoma40. The gene CTTN lies just downstream of the smallest region of overlap of the four cell lines, however, three of the four amplifications include CTTN (SCCNC2, 6 and 7).
TP53, EGFR, KRAS and CDKN2A (encoding p16) are genes well studied in the literature on sinonasal SCC. TP53 mutation and p53 overexpression have been reported in 70% and 50% of tumors, respectively41,42,43. EGFR overexpression has been found in about 40%43,44 and p16 immunopositivity in 21% of cases8,9. Mutations in KRAS are very rare, occurring in less than 1%12,45. These genetic characteristics are reflected by our six cell lines. We found TP53 mutation in three and p53 overexpression p53 in five of the cell lines. EGFR expression was observed in four cell lines and KRAS or BRAF mutations were absent. Not in agreement with findings in the literature was the p16 immunonegative staining in the cell lines. One primary tumor was weakly positive (SCCNC4), but not its derivative cell line. HPV analysis was negative for all cell line and primary tumor samples, which is in agreement with the absence of strong p16 immunopositivity46. It has been suggested that approximately 15–20% of sinonasal SCC are HPV-positive and may have developed from inverted papilloma9. It is unfortunate that none of our cell lines represent this subset of sinonasal SCC. We did however have one cell line carrying with few aberrations, representing a subset of tumors with a near-diploid karyotype. For head and neck SCC in general it has been estimated that 15% of all tumors are diploid47.
We have positively identified each cell line as derived from its corresponding patient by short tandem repeat fingerprinting, demonstrating at least 90% identical repeats. The fact that there was no 100% match reflects the genomic imbalances present in the tumor. Extra confirmation of the uniqueness and identity of the cell lines was obtained from the similarity between the microarray CGH profiles of the cell lines and the primary tumors from which they were derived. The lower amplitude of the gains and losses in the primary tumors can be adscribed to contamination of the sample with normals cells such as stroma and infiltrating lymphocytes. Nevertheless, in every cell line there were 2–3 chromosomes (11 in SCCNC7) carrying copy number changes that could not be observed in the primary tumor. It may be that these ‘extra’ abnormalities have been acquired during the time of establishment. Another possibility is that the final established cell line is the result of an selection process of a minor subclone within a genetically heterogeneous tumor that best adapted to the in vitro growth conditions. There were no specific ‘extra’ aberrations occuring in more than one cell line that could suggest specific genes or pathways implicated in or required for in vitro growth capacity.
In conclusion, this paper describes six unique cell lines derived from previously untreated, primary sinonasal squamous cell carcinoma. We have shown that they are representative of sinonasal SCC with regard to both their clinical and genetic diversity and we have shown differences in proliferative and invasive characteristics in vitro. Together with the high resolution genetic data obtained, these cell lines will be a useful tool for preclinical testing of new therapeutic agents.
Additional information
Sources of support: Grant PI05-1387, PI08-1599 and EMER07-048 of Fondos de Investigación Sanitaria (FIS) and RD12/0036/0015 of Red Temática de Investigación Cooperativa en Cáncer (RTICC), Spain.
References
Ansa, B. et al. Paranasal sinus squamous cell carcinoma incidence and survival based on Surveillance, Epidemiology and End Results data, 1973 to 2009. Cancer. 119, 2602–2610 (2013).
Turner, J. H. & Reh, D. D. Incidence and survival in patients with sinonasal cancer: a historical analysis of population-based data. Head Neck. 34, 877–885 (2012).
Barnes, L. Pathology and Genetics of Head and Neck Tumours. World Health Organization Classification of Tumours, vol. 9. (IARC Press, Lyon, 2005).
Bonzini, M. et al. Prevalence of occupational hazards in patients with different types of epithelial sinonasal cancers. Rhinology. 51, 31–36 (2013).
Sanghvi, S. et al. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4,994 patients. Laryngoscope. 124, 76–83 (2014).
IARC. Monographs on the Evaluation of Carcinogenic Risks to Humans. Tobacco smoke and involuntary smoking. (IARC Press, Lyon, 2004).
Youlden, D. R. et al. International comparisons of the incidence and mortality of sinonasal cancer. Cancer Epidemiol. 37, 770–779 (2013).
Alos, L. et al. Human papillomaviruses are identified in a subgroup of sinonasal squamous cell carcinomas with favorable outcome. Cancer. 115, 2701–2709 (2009).
Bishop, J. A. et al. Human papillomavirus-related carcinomas of the sinonasal tract. Am. J. Surg. Pathol. 37, 185–192 (2013).
Dulguerov, P. & Allal, A. S. Nasal and paranasal sinus carcinoma: how can we continue to make progress? Curr. Opin. Otolaryngol. Head Neck Surg. 14, 67–72 (2006).
Sobin, L. H., Gospodarowicz, M. K. & Wittekind, C. H. eds. International Union Against Cancer (UICC) TNM Classification of Malignant Tumors. 7th ed. (Wiley-Blackwell, Oxford, 2009).
Rodrigo, J. P. et al. Time trends in the prevalence of HPV in oropharyngeal squamous cell carcinomas in northern Spain (1990–2009). Int. J. Cancer. 134, 487–92 (2014).
Pérez-Escuredo, J. et al. Wood dust-related mutational profile of TP53 in intestinal-type sinonasal adenocarcinoma. Hum. Pathol. 43, 1894–901 (2012).
López, F. et al. KRAS and BRAF mutations in sinonasal cancer. Oral Oncol. 48, 692–697 (2012).
Álvarez-Argüelles, M. E. et al. Human papillomavirus infection in a male population attending a sexually transmitted infection service. PLoS One. 8, e54375 (2013).
Pérez-Escuredo, J. et al. Establishment and genetic characterization of an immortal tumor cell line derived from intestinal-type sinonasal adenocarcinoma. Cell. Oncol. (Dordr). 34, 23–31 (2011).
Takahashi, Y. et al. Establishment and characterization of novel cell lines from sinonasal undifferentiated carcinoma. Clin. Cancer Res. 18, 6178–6187 (2012).
Noguchi, K. et al. Establishment of a new cell line with neuronal differentiation derived from small cell neuroendocrine carcinoma of the maxillary sinus. Oncology. 66, 234–243 (2004).
Kishimoto, H. et al. Isolation and characterisation of adenoid squamous carcinoma cells highly producing SCC antigen and CEA from carcinoma of the maxillary sinus. Oral Oncol. 36, 70–75 (2000).
Sorensen, P. H. et al. Olfactory neuroblastoma is a peripheral primitive neuroectodermal tumor related to Ewing sarcoma. Proc. Natl. Acad. Sci. U S A. 93, 1038–1043 (1996).
Krause, C. J. et al. Human squamous cell carcinoma. Establishment and characterization of new permanent cell lines. Arch. Otolaryngol. 107, 703–710 (1981).
Lin, C. J. et al. Head and neck squamous cell carcinoma cell lines: established models and rationale for selection. Head Neck. 29, 163–188 (2007).
Brenner, J. C. et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 32, 417–426 (2010).
Zhao, M. et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin. Cancer Res. 17, 7248–7264 (2011).
Moore, G. E. & Sandberg, A. A. Studies of a human tumor cell line with a diploid karyotype. Cancer. 17, 170–175 (1964).
Liebertz, D. J. et al. Establishment and characterization of a novel head and neck squamous cell carcinoma cell line USC-HN1. Head Neck Oncol. 22, 2–5 (2010).
Kudoh, S. et al. Appearance of drug-resistant tumor cells in a maxillary cancer patient undergoing chemotherapy and chemoradiotherapy. Am. J. Otolaryngol. 11, 165–169 (1990).
Komiyama, S. et al. Establishment of tumor cell lines from a patient with head and neck cancer and their different sensitivities to anti-cancer agents. Cancer. 63, 675–681 (1989).
Lansford, C. et al. Head and neck cancers. In: Human cell culture, Vol 2, cancer cell lines, Part 2. (ed. Masters, J. R. & Palsson, B.) 185–255 (Kluwer Academic Publishers, Dordrecht, 1999).
Arosarena, O. A. et al. Expression of major histocompatibility complex antigens in squamous cell carcinomas of the head and neck: effects of interferon gene transfer. Otolaryngol. Head Neck Surg. 120, 665–671 (1999).
Kim, S. Y. et al. Establishment and characterization of nine new head and neck cancer cell lines. Acta Otolaryngol. 117, 775–784 (1997).
Nakashima, T., Makishima, K. & Hiroto, I. Establishment of a new cell line from maxillary sinus carcinoma. Ann. Otol. Rhinol. Laryngol. 89, 24–8 (1980).
Komiyama, S. et al. Heterogeneity in epidermal growth factor responsiveness and tumor growth of human maxillary cancer cell lines. Ann. Otol. Rhinol. Laryngol. 101, 519–24 (1992).
Fukiage, T. et al. Establishment of a human cell line from maxillary squamous cell carcinoma and its biological features as a stimulator for induction of cytotoxic T lymphocytes. Auris Nasus Larynx. 21, 163–172 (1994).
López, F. et al. Genomic profiling of sinonasal squamous cell carcinoma. Head Neck. 33, 145–153 (2011).
Varshavsky, A. Targeting the absence: Homozygous DNA deletions as immutable signposts for cancer therapy. Proc. Natl. Acad. Sci. USA. 104, 14935–14940 (2007).
Pattje, W. J. et al. FADD expression is associated with regional and distant metastasis in squamous cell carcinoma of the head and neck. Histopathology. 63, 263–70 (2013).
Rodrigo, J. P. et al. Distinctive clinicopathological associations of amplification of the cortactin gene at 11q13 in head and neck squamous cell carcinomas. J. Pathol. 217, 516–23 (2009).
Wilkerson, P. M. & Reis-Filho, J. S. The 11q13–q14 amplicon: clinicopathological correlations and potential drivers. Genes Chromosomes Cancer. 52, 333–55 (2013).
Rodrigo, J. P. et al. Cortactin and focal adhesion kinase as predictors of cancer risk in patients with laryngeal premalignancy. Cancer Prev. Res. (Phila). 4, 1333–41 (2011).
Holmila, R. et al. COX-2 and p53 in human sinonasal cancer: COX-2 expression is associated with adenocarcinoma histology and wood-dust exposure. Int. J. Cancer. 122, 2154–2159 (2008).
Holmila, R. et al. Mutations in TP53 tumor suppressor gene in wood dust-related sinonasal cancer. Int. J. Cancer. 127, 578–588 (2010).
Takahashi, Y. et al. Comprehensive assessment of prognostic markers for sinonasal squamous cell carcinoma. Head Neck. 10.1002/hed.23423. [Epub ahead of print] (2013).
López, F. et al. Gene amplification and protein overexpression of EGFR and ERBB2 in sinonasal squamous cell carcinoma. Cancer. 118, 1818–1826 (2012).
Bornholdt, J. et al. K-ras mutations in sinonasal cancers in relation to wood dust exposure. BMC Cancer. 8, 53 (2008).
El-Naggar, A. K. & Westra, W. H. p16 expression as a surrogate marker for HPV-related oropharyngeal carcinoma: a guide for interpretative relevance and consistency. Head Neck. 34, 459–461 (2012).
Leemans, C. R., Braakhuis, B. J. & Brakenhoff, R. H. The molecular biology of head and neck cancer. Nat. Rev. Cancer. 11, 9–22 (2011).
Acknowledgements
Our thanks to Dr. Milagros Balbín of the department of Molecular Oncology at the Hospital Universitario Central de Asturias for the STR genotyping and Dr. Blanca Vivanco for the pathological evaluation.
Author information
Authors and Affiliations
Contributions
J.L.L. and F.L. operated the patients and provided the tissue material and the clinical data. M.A.G. and S.P. initiated and maintained the cell lines. C.G.I. and S.P. performed the proliferation and invasion assays and the mutation analysis. C.G.I. and E.A. carried out the immunohistochemistry and immunofluorescence experiments. A.L.H. and M.A.H. were responsible for the microarray CGH analysis and SM for the HPV analysis. M.A.H., J.L.L. and F.L. performed the literature search and the extraction of all relevant clinical data of previously published sinonasal cell lines shown in table 3. M.A.H. and C.G.I. wrote the main manuscript text and prepared the figures. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
García-Inclán, C., López-Hernández, A., Alonso-Guervós, M. et al. Establishment and genetic characterization of six unique tumor cell lines as preclinical models for sinonasal squamous cell carcinoma. Sci Rep 4, 4925 (2014). https://doi.org/10.1038/srep04925
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04925
This article is cited by
-
Experimental Models of Sinonasal Tumors for Preclinical Testing of Candidate Targeted Therapies
Current Otorhinolaryngology Reports (2023)
-
Establishment and genetic characterization of cell lines derived from proliferating nasal polyps and sinonasal inverted papillomas
Scientific Reports (2021)
-
Sinonasal carcinoma: clinical, pathological, genetic and therapeutic advances
Nature Reviews Clinical Oncology (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
